
Cell Type-Specific Promoters of Volvox carteri for Molecular Cell Biology Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. MA-Plot (Bland–Altman Plot)
2.2. Strains and Culture Conditions
2.3. PCR Amplification of DNA Fragments for Vector Construction
2.4. Construction of Vectors for Cell Type-Specific Expression of Luciferase
2.5. Stable Nuclear Transformation of V. carteri by Particle Bombardment
2.6. Genomic PCR
2.7. Fast Luciferase Screening Assay
2.8. Separation of the Cell Types of V. carteri
2.9. Isolation of Total RNA from Separated Cell Types
2.10. Quantitative Real-Time RT-PCR
2.11. Quantification of Luciferase Activity in Separated Cell Types
2.12. Determination of the Chlorophyll Concentration
3. Results
3.1. Identification of Suitable Genes with Strong Cell Type-Specific Expression
3.2. Construction of Chimeric Genes for Promoter Analysis and Application
3.3. Generation of Stable Transgenic Volvox Strains with Chimeric Genes
3.4. Fast Luciferase Screening Assay for General Detection of Transgene Expression
3.5. Cell Type-Specific mRNA Expression of Transgenes Driven by PCY1 and PFP Promoters
3.6. Quantification of PCY1- or PFP-Regulated Luciferase Activity Separately for Each Cell Type
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kirk, D.L. Volvox: Molecular-Genetic Origins of Multicellularity and Cellular Differentiation, 1st ed.; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar]
- Hallmann, A. Evolution of reproductive development in the volvocine algae. Sex. Plant Reprod. 2011, 24, 97–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nematollahi, G.; Kianianmomeni, A.; Hallmann, A. Quantitative analysis of cell-type specific gene expression in the green alga Volvox carteri. BMC Genom. 2006, 7, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, B.; Wibberg, D.; Hallmann, A. Whole transcriptome RNA-Seq analysis reveals extensive cell type-specific compartmentalization in Volvox carteri. BMC Biol. 2017, 15, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matt, G.Y.; Umen, J.G. Cell-type transcriptomes of the multicellular green alga Volvox carteri yield insights into the evolutionary origins of germ and somatic differentiation programs. G3 2018, 8, 531–550. [Google Scholar] [CrossRef] [Green Version]
- Day, T.C.; Höhn, S.S.; Zamani-Dahaj, S.A.; Yanni, D.; Burnetti, A.; Pentz, J.; Honerkamp-Smith, A.R.; Wioland, H.; Sleath, H.R.; Ratcliff, W.C.; et al. Cellular organization in lab-evolved and extant multicellular species obeys a maximum entropy law. eLife 2022, 11, e72707. [Google Scholar] [CrossRef]
- Matt, G.; Umen, J. Volvox: A simple algal model for embryogenesis, morphogenesis and cellular differentiation. Dev. Biol. 2016, 419, 99–113. [Google Scholar] [CrossRef]
- Lindsey, C.R.; Rosenzweig, F.; Herron, M.D. Phylotranscriptomics points to multiple independent origins of multicellularity and cellular differentiation in the volvocine algae. BMC Biol. 2021, 19, 182. [Google Scholar] [CrossRef]
- Kirk, D.L. A twelve-step program for evolving multicellularity and a division of labor. BioEssays 2005, 27, 299–310. [Google Scholar] [CrossRef]
- Tian, Y.; Gao, S.; von der Heyde, E.L.; Hallmann, A.; Nagel, G. Two-component cyclase opsins of green algae are ATP-dependent and light-inhibited guanylyl cyclases. BMC Biol. 2018, 16, 144. [Google Scholar] [CrossRef] [Green Version]
- Kianianmomeni, A.; Hallmann, A. Transcriptional analysis of Volvox photoreceptors suggests the existence of different cell-type specific light-signaling pathways. Curr. Genet. 2015, 61, 3–18. [Google Scholar] [CrossRef]
- Kianianmomeni, A.; Stehfest, K.; Nematollahi, G.; Hegemann, P.; Hallmann, A. Channelrhodopsins of Volvox carteri are photochromic proteins that are specifically expressed in somatic cells under control of light, temperature, and the sex inducer. Plant Physiol. 2009, 151, 347–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueki, N.; Matsunaga, S.; Inouye, I.; Hallmann, A. How 5000 independent rowers coordinate their strokes in order to row into the sunlight: Phototaxis in the multicellular green alga Volvox. BMC Biol. 2010, 8, 103. [Google Scholar] [CrossRef] [Green Version]
- Drescher, K.; Goldstein, R.E.; Tuval, I. Fidelity of adaptive phototaxis. Proc. Natl. Acad. Sci. USA 2010, 107, 11171–11176. [Google Scholar] [CrossRef] [PubMed]
- von der Heyde, E.L.; Hallmann, A. Molecular and cellular dynamics of early embryonic cell divisions in Volvox carteri. Plant Cell 2022, 34, 1326–1353. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Pappas, V.; Hallmann, A.; Miller, S.M. Hsp70A and GlsA interact as partner chaperones to regulate asymmetric division in Volvox. Dev. Biol. 2005, 286, 537–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumper, M.; Hallmann, A. Biochemistry of the extracellular matrix of Volvox. Int. Rev. Cytol. 1998, 180, 51–85. [Google Scholar]
- Hallmann, A. The pherophorins: Common, versatile building blocks in the evolution of extracellular matrix architecture in Volvocales. Plant J. 2006, 45, 292–307. [Google Scholar] [CrossRef]
- Nishimura, M.; Nagashio, R.; Sato, Y.; Hasegawa, T. Late Somatic Gene 2 disrupts parental spheroids cooperatively with Volvox hatching enzyme A in Volvox. Planta 2017, 245, 183–192. [Google Scholar] [CrossRef]
- von der Heyde, B.; Hallmann, A. Targeted migration of pherophorin-S indicates extensive extracellular matrix dynamics in Volvox carteri. Plant J. 2020, 103, 2301–2317. [Google Scholar] [CrossRef]
- von der Heyde, B.; Hallmann, A. Cell type-specific pherophorins of Volvox carteri reveal interplay of both cell types in ECM biosynthesis. Cells 2023, 12, 134. [Google Scholar] [CrossRef]
- Viamontes, G.I.; Kirk, D.L. Cell shape changes and the mechanism of inversion in Volvox. J. Cell Biol. 1977, 75, 719–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirk, D.L.; Viamontes, G.I.; Green, K.J.; Bryant, J.L., Jr. Integrated morphogenetic behavior of cell sheets: Volvox as a model. In Developmental Order: Its Origin and Regulation; Subtelny, S., Green, P.B., Eds.; Alan R. Liss: New York, NY, USA, 1982; pp. 247–274. [Google Scholar]
- Kirk, D.L.; Nishii, I. Volvox carteri as a model for studying the genetic and cytological control of morphogenesis. Dev. Growth Differ. 2001, 43, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Nishii, I.; Ogihara, S.; Kirk, D.L. A kinesin, invA, plays an essential role in Volvox morphogenesis. Cell 2003, 113, 743–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallmann, A. Morphogenesis in the family Volvocaceae: Different tactics for turning an embryo right-side out. Protist 2006, 157, 445–461. [Google Scholar] [CrossRef]
- Haas, P.A.; Goldstein, R.E. Embryonic inversion in Volvox carteri: The flipping and peeling of elastic lips. Phys. Rev. E 2018, 98, 052415. [Google Scholar] [CrossRef] [Green Version]
- Prochnik, S.E.; Umen, J.; Nedelcu, A.M.; Hallmann, A.; Miller, S.M.; Nishii, I.; Ferris, P.; Kuo, A.; Mitros, T.; Fritz-Laylin, L.K.; et al. Genomic analysis of organismal complexity in the multicellular green alga Volvox carteri. Science 2010, 329, 223–226. [Google Scholar] [CrossRef] [Green Version]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Marechal-Drouard, L.; et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef] [Green Version]
- Craig, R.J.; Gallaher, S.D.; Shu, S.; Salome, P.; Jenkins, J.W.; Blaby-Haas, C.E.; Purvine, S.O.; O’Donnell, S.; Barry, K.; Grimwood, J.; et al. The Chlamydomonas Genome Project, version 6: Reference assemblies for mating type plus and minus strains reveal extensive structural mutation in the laboratory. Plant Cell 2023, 35, 644–672. [Google Scholar] [CrossRef]
- Blaby, I.K.; Blaby-Haas, C.E.; Tourasse, N.; Hom, E.F.; Lopez, D.; Aksoy, M.; Grossman, A.; Umen, J.; Dutcher, S.; Porter, M.; et al. The Chlamydomonas genome project: A decade on. Trends Plant Sci. 2014, 19, 672–680. [Google Scholar] [CrossRef] [Green Version]
- Featherston, J.; Arakaki, Y.; Hanschen, E.R.; Ferris, P.J.; Michod, R.E.; Olson, B.; Nozaki, H.; Durand, P.M. The 4-celled Tetrabaena socialis nuclear genome reveals the essential components for genetic control of cell number at the origin of multicellularity in the volvocine lineage. Mol. Biol. Evol. 2018, 35, 855–870. [Google Scholar] [CrossRef]
- Hanschen, E.R.; Marriage, T.N.; Ferris, P.J.; Hamaji, T.; Toyoda, A.; Fujiyama, A.; Neme, R.; Noguchi, H.; Minakuchi, Y.; Suzuki, M.; et al. The Gonium pectorale genome demonstrates co-option of cell cycle regulation during the evolution of multicellularity. Nat. Commun. 2016, 7, 11370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, S.; Yamamoto, K.; Matsuzaki, R.; Suzuki, S.; Yamaguchi, H.; Hirooka, S.; Minakuchi, Y.; Miyagishima, S.Y.; Kawachi, M.; Toyoda, A.; et al. Genome sequencing of the multicellular alga Astrephomene provides insights into convergent evolution of germ-soma differentiation. Sci. Rep. 2021, 11, 22231. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Marin, B.; Rakijas, J.B.; Tyagi, A.; Pandey, A.; Hanschen, E.R.; Anderson, J.; Heffel, M.G.; Platt, T.G.; Olson, B. Gene loss during a transition to multicellularity. Sci. Rep. 2023, 13, 5268. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Ostell, J.; Pruitt, K.D.; Sayers, E.W. GenBank. Nucleic Acids Res. 2018, 46, D41–D47. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Ostell, J.; Pruitt, K.D.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2020, 48, D84–D86. [Google Scholar] [CrossRef] [Green Version]
- Dueck, A.; Evers, M.; Henz, S.R.; Unger, K.; Eichner, N.; Merkl, R.; Berezikov, E.; Engelmann, J.C.; Weigel, D.; Wenzl, S.; et al. Gene silencing pathways found in the green alga Volvox carteri reveal insights into evolution and origins of small RNA systems in plants. BMC Genom. 2016, 17, 853. [Google Scholar] [CrossRef] [Green Version]
- Kianianmomeni, A.; Ong, C.S.; Rätsch, G.; Hallmann, A. Genome-wide analysis of alternative splicing in Volvox carteri. BMC Genom. 2014, 15, 1117. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, R.N.; Umen, J. Identification of cell-type specific alternative transcripts in the multicellular alga Volvox carteri. bioRxiv 2023. [Google Scholar] [CrossRef]
- Schiedlmeier, B.; Schmitt, R.; Müller, W.; Kirk, M.M.; Gruber, H.; Mages, W.; Kirk, D.L. Nuclear transformation of Volvox carteri. Proc. Natl. Acad. Sci. USA 1994, 91, 5080–5084. [Google Scholar] [CrossRef] [PubMed]
- Hallmann, A.; Rappel, A.; Sumper, M. Gene replacement by homologous recombination in the multicellular green alga Volvox carteri. Proc. Natl. Acad. Sci. USA 1997, 94, 7469–7474. [Google Scholar] [CrossRef] [PubMed]
- Gruber, H.; Goetinck, S.D.; Kirk, D.L.; Schmitt, R. The nitrate reductase-encoding gene of Volvox carteri: Map location, sequence and induction kinetics. Gene 1992, 120, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Hallmann, A.; Rappel, A. Genetic engineering of the multicellular green alga Volvox: A modified and multiplied bacterial antibiotic resistance gene as a dominant selectable marker. Plant J. 1999, 17, 99–109. [Google Scholar] [CrossRef]
- Hallmann, A.; Sumper, M. Reporter genes and highly regulated promoters as tools for transformation experiments in Volvox carteri. Proc. Natl. Acad. Sci. USA 1994, 91, 11562–11566. [Google Scholar] [CrossRef]
- Hallmann, A.; Sumper, M. The Chlorella hexose/H+ symporter is a useful selectable marker and biochemical reagent when expressed in Volvox. Proc. Natl. Acad. Sci. USA 1996, 93, 669–673. [Google Scholar] [CrossRef]
- Jakobiak, T.; Mages, W.; Scharf, B.; Babinger, P.; Stark, K.; Schmitt, R. The bacterial paromomycin resistance gene, aphH, as a dominant selectable marker in Volvox carteri. Protist 2004, 155, 381–393. [Google Scholar] [CrossRef]
- Ortega-Escalante, J.A.; Kwok, O.; Miller, S.M. New selectable markers for Volvox carteri transformation. Protist 2019, 170, 52–63. [Google Scholar] [CrossRef]
- Pappas, V.; Miller, S.M. Functional analysis of the Volvox carteri asymmetric division protein GlsA. Mech. Dev. 2009, 126, 842–851. [Google Scholar] [CrossRef]
- Ender, F.; Godl, K.; Wenzl, S.; Sumper, M. Evidence for autocatalytic cross-linking of hydroxyproline-rich glycoproteins during extracellular matrix assembly in Volvox. Plant Cell 2002, 14, 1147–1160. [Google Scholar] [CrossRef] [Green Version]
- von der Heyde, E.L.; Klein, B.; Abram, L.; Hallmann, A. The inducible nitA promoter provides a powerful molecular switch for transgene expression in Volvox carteri. BMC Biotechnol. 2015, 15, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von der Heyde, E.L.; Hallmann, A. Babo1, formerly Vop1 and Cop1/2, is no eyespot photoreceptor but a basal body protein illuminating cell division in Volvox carteri. Plant J. 2020, 102, 276–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirk, M.M.; Stark, K.; Miller, S.M.; Müller, W.; Taillon, B.E.; Gruber, H.; Schmitt, R.; Kirk, D.L. regA, a Volvox gene that plays a central role in germ-soma differentiation, encodes a novel regulatory protein. Development 1999, 126, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.M.; Kirk, D.L. glsA, a Volvox gene required for asymmetric division and germ cell specification, encodes a chaperone-like protein. Development 1999, 126, 649–658. [Google Scholar] [CrossRef]
- Ueki, N.; Nishii, I. Idaten is a new cold-inducible transposon of Volvox carteri that can be used for tagging developmentally important genes. Genetics 2008, 180, 1343–1353. [Google Scholar] [CrossRef] [Green Version]
- Geng, S.; De Hoff, P.; Umen, J.G. Evolution of sexes from an ancestral mating-type specification pathway. PLoS Biol. 2014, 12, e1001904. [Google Scholar] [CrossRef]
- Ortega-Escalante, J.A.; Jasper, R.; Miller, S.M. CRISPR/Cas9 mutagenesis in Volvox carteri. Plant J. 2019, 97, 661–672. [Google Scholar] [CrossRef] [Green Version]
- Godl, K.; Hallmann, A.; Wenzl, S.; Sumper, M. Differential targeting of closely related ECM glycoproteins: The pherophorin family from Volvox. EMBO J. 1997, 16, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Hallmann, A.; Sumper, M. An inducible arylsulfatase of Volvox carteri with properties suitable for a reporter-gene system. Purification, characterization and molecular cloning. Eur. J. Biochem. 1994, 221, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Hilker, R.; Stadermann, K.B.; Doppmeier, D.; Kalinowski, J.; Stoye, J.; Straube, J.; Winnebald, J.; Goesmann, A. ReadXplorer—Visualization and analysis of mapped sequences. Bioinformatics 2014, 30, 2247–2254. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; McCarthy, D.J.; Chen, Y.; Okoniewski, M.; Smyth, G.K.; Huber, W.; Robinson, M.D. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 2013, 8, 1765–1786. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016; Available online: https://www.R-project.org/ (accessed on 15 May 2023).
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Society. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Starr, R.C. Structure, reproduction and differentiation in Volvox carteri f. nagariensis Iyengar, strains HK 9 & 10. Arch. Protistenkd. 1969, 111, 204–222. [Google Scholar]
- Starr, R.C. Control of differentiation in Volvox. Dev. Biol. Suppl. 1970, 4, 59–100. [Google Scholar]
- Kianianmomeni, A.; Nematollahi, G.; Hallmann, A. A gender-specific retinoblastoma-related protein in Volvox carteri implies a role for the retinoblastoma protein family in sexual development. Plant Cell 2008, 20, 2399–2419. [Google Scholar] [CrossRef] [Green Version]
- Provasoli, L.; Pintner, I.J. Artificial media for fresh-water algae: Problems and suggestions. In The Ecology of Algae, a Symposium Held at the Pymatuning Laboratory of Field Biology on 18–19 June 1959, 1st ed.; Tryon, C.A., Hartman, R.T., Eds.; The Pymatuning Symposia in Ecology, Special Publication No. 2; University of Pittsburgh: Pittsburgh, PA, USA, 1959; pp. 84–96. [Google Scholar]
- Starr, R.C.; Jaenicke, L. Purification and characterization of the hormone initiating sexual morphogenesis in Volvox carteri f. nagariensis Iyengar. Proc. Natl. Acad. Sci. USA 1974, 71, 1050–1054. [Google Scholar] [CrossRef]
- Edwards, K.; Johnstone, C.; Thompson, C. A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res. 1991, 19, 1349. [Google Scholar] [CrossRef]
- Shao, N.; Bock, R. A codon-optimized luciferase from Gaussia princeps facilitates the in vivo monitoring of gene expression in the model alga Chlamydomonas reinhardtii. Curr. Genet. 2008, 53, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Hallmann, A.; Wodniok, S. Swapped green algal promoters: aphVIII-based gene constructs with Chlamydomonas flanking sequences work as dominant selectable markers in Volvox and vice versa. Plant Cell Rep. 2006, 25, 582–591. [Google Scholar] [CrossRef]
- Lerche, K.; Hallmann, A. Stable nuclear transformation of Eudorina elegans. BMC Biotechnol. 2013, 13, 11. [Google Scholar] [CrossRef] [Green Version]
- Lerche, K.; Hallmann, A. Stable nuclear transformation of Gonium pectorale. BMC Biotechnol. 2009, 9, 64. [Google Scholar] [CrossRef] [Green Version]
- Gruber, H.; Kirzinger, S.H.; Schmitt, R. Expression of the Volvox gene encoding nitrate reductase: Mutation-dependent activation of cryptic splice sites and intron-enhanced gene expression from a cDNA. Plant Mol. Biol. 1996, 31, 1–12. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerche, K.; Hallmann, A. Stable nuclear transformation of Pandorina morum. BMC Biotechnol. 2014, 14, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siaut, M.; Heijde, M.; Mangogna, M.; Montsant, A.; Coesel, S.; Allen, A.; Manfredonia, A.; Falciatore, A.; Bowler, C. Molecular toolbox for studying diatom biology in Phaeodactylum tricornutum. Gene 2007, 406, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Kianianmomeni, A.; Hallmann, A. Validation of reference genes for quantitative gene expression studies in Volvox carteri using real-time RT-PCR. Mol. Biol. Rep. 2013, 40, 6691–6699. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 2000, 25, 169–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Jeffrey, S.W.; Humphrey, G.F. New spectrophotometric equations for determining chlorophyll a, b, c1 and c2 in higher plants, algae and natural phytoplankton. Biochem. Physiol. Pflanz. 1975, 167, 191–194. [Google Scholar] [CrossRef]
- Barnes, A.T.; Case, J.F. Bioluminescence in the mesopelagic copepod, Gaussia princeps (T. Scott). J. Exp. Mar. Biol. Ecol. 1972, 8, 53–71. [Google Scholar] [CrossRef]
- Szent-Gyorgyi, C.; Ballou, B.T.; Dagmal, E.; Bryan, B. Cloning and characterization of new bioluminescent proteins. Proc. SPIE 1999, 3600, 4–11. [Google Scholar]
- Neupert, J.; Gallaher, S.D.; Lu, Y.; Strenkert, D.; Segal, N.; Barahimipour, R.; Fitz-Gibbon, S.T.; Schroda, M.; Merchant, S.S.; Bock, R. An epigenetic gene silencing pathway selectively acting on transgenic DNA in the green alga Chlamydomonas. Nat. Commun. 2020, 11, 6269. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, H.; Johnson, A.M.; Gillham, N.W.; Boynton, J.E. Epigenetic silencing of a foreign gene in nuclear transformants of Chlamydomonas. Plant Cell 1997, 9, 925–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, S.L.; Warkentin, T.D.; DeLong, C.M. Transgene copy number can be positively or negatively associated with transgene expression. Plant Mol. Biol. 1993, 21, 17–26. [Google Scholar] [CrossRef]
- Cabrera, A.; Edelstein, H.I.; Glykofrydis, F.; Love, K.S.; Palacios, S.; Tycko, J.; Zhang, M.; Lensch, S.; Shields, C.E.; Livingston, M.; et al. The sound of silence: Transgene silencing in mammalian cell engineering. Cell Syst. 2022, 13, 950–973. [Google Scholar] [CrossRef]
- Jones, J.D.; Dunsmuir, P.; Bedbrook, J. High level expression of introduced chimaeric genes in regenerated transformed plants. EMBO J. 1985, 4, 2411–2418. [Google Scholar] [CrossRef]
- Peach, C.; Velten, J. Transgene expression variability (position effect) of CAT and GUS reporter genes driven by linked divergent T-DNA promoters. Plant Mol. Biol. 1991, 17, 49–60. [Google Scholar] [CrossRef]
- Yamasaki, T.; Ohama, T. Involvement of Elongin C in the spread of repressive histone modifications. Plant J. 2011, 65, 51–61. [Google Scholar] [CrossRef]
- Schoppmeier, J.; Mages, W.; Lechtreck, K.F. GFP as a tool for the analysis of proteins in the flagellar basal apparatus of Chlamydomonas. Cell Motil. Cytoskelet. 2005, 61, 189–200. [Google Scholar] [CrossRef]
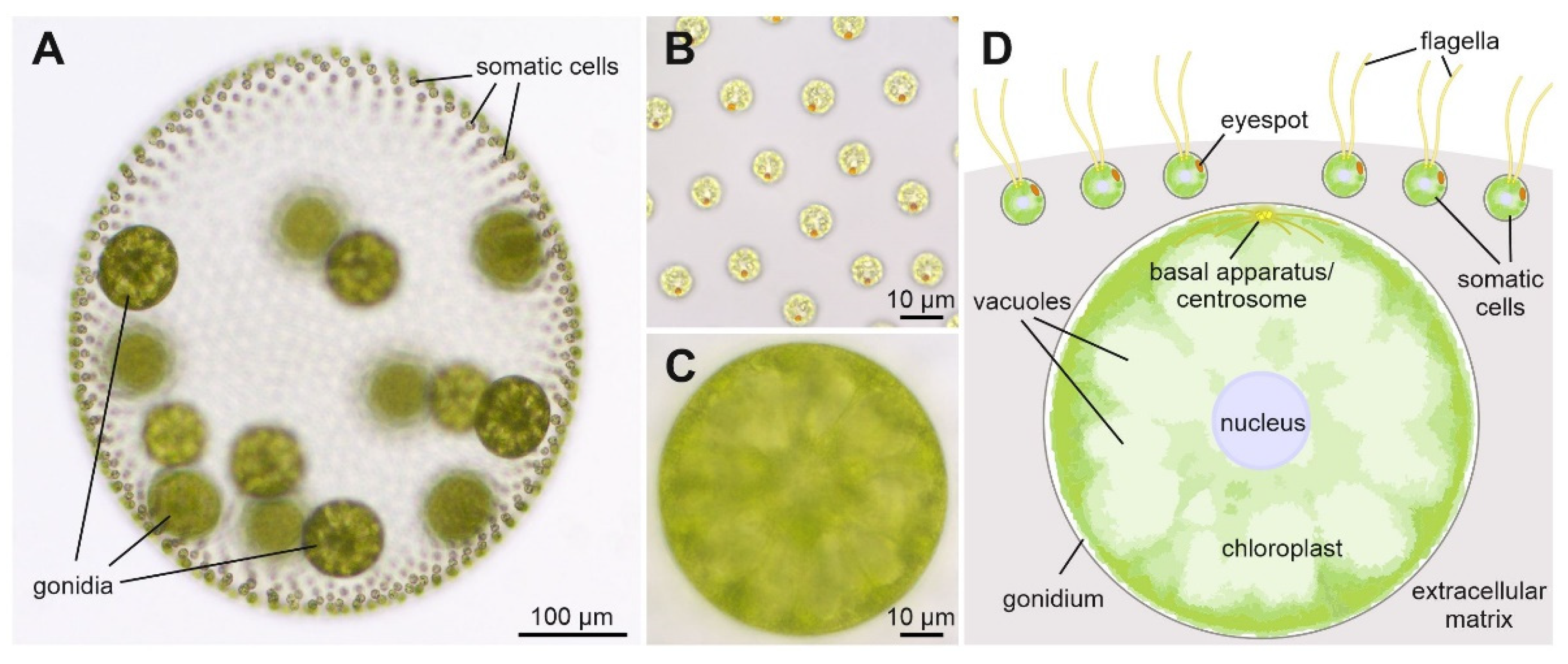





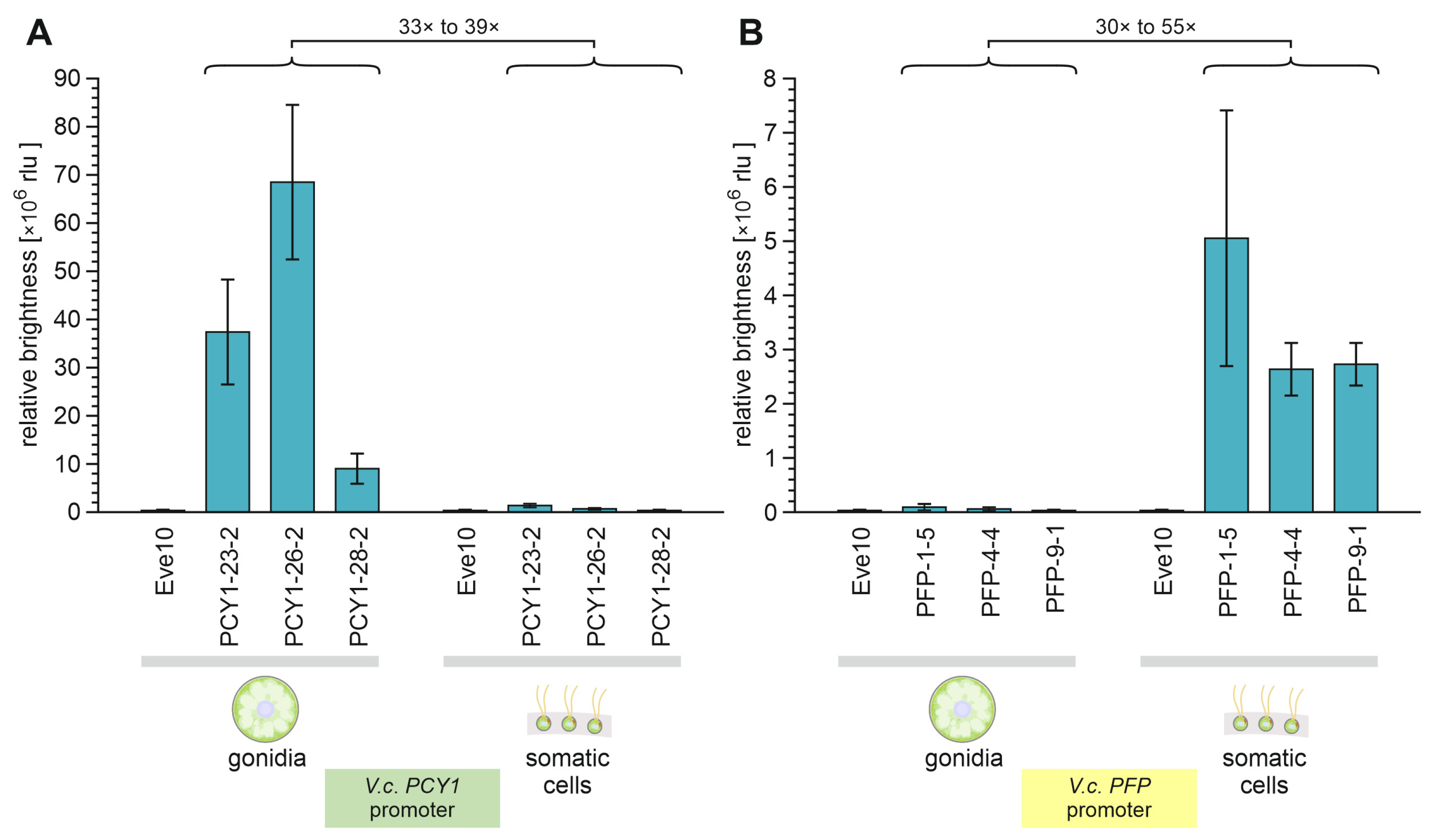
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
von der Heyde, B.; von der Heyde, E.L.; Hallmann, A. Cell Type-Specific Promoters of Volvox carteri for Molecular Cell Biology Studies. Genes 2023, 14, 1389. https://doi.org/10.3390/genes14071389
von der Heyde B, von der Heyde EL, Hallmann A. Cell Type-Specific Promoters of Volvox carteri for Molecular Cell Biology Studies. Genes. 2023; 14(7):1389. https://doi.org/10.3390/genes14071389
Chicago/Turabian Stylevon der Heyde, Benjamin, Eva Laura von der Heyde, and Armin Hallmann. 2023. "Cell Type-Specific Promoters of Volvox carteri for Molecular Cell Biology Studies" Genes 14, no. 7: 1389. https://doi.org/10.3390/genes14071389
APA Stylevon der Heyde, B., von der Heyde, E. L., & Hallmann, A. (2023). Cell Type-Specific Promoters of Volvox carteri for Molecular Cell Biology Studies. Genes, 14(7), 1389. https://doi.org/10.3390/genes14071389