
k-mer-Based Genome-Wide Association Studies in Plants: Advances, Challenges, and Perspectives


Abstract
:1. Use of Genome-Wide Association Studies in Crops
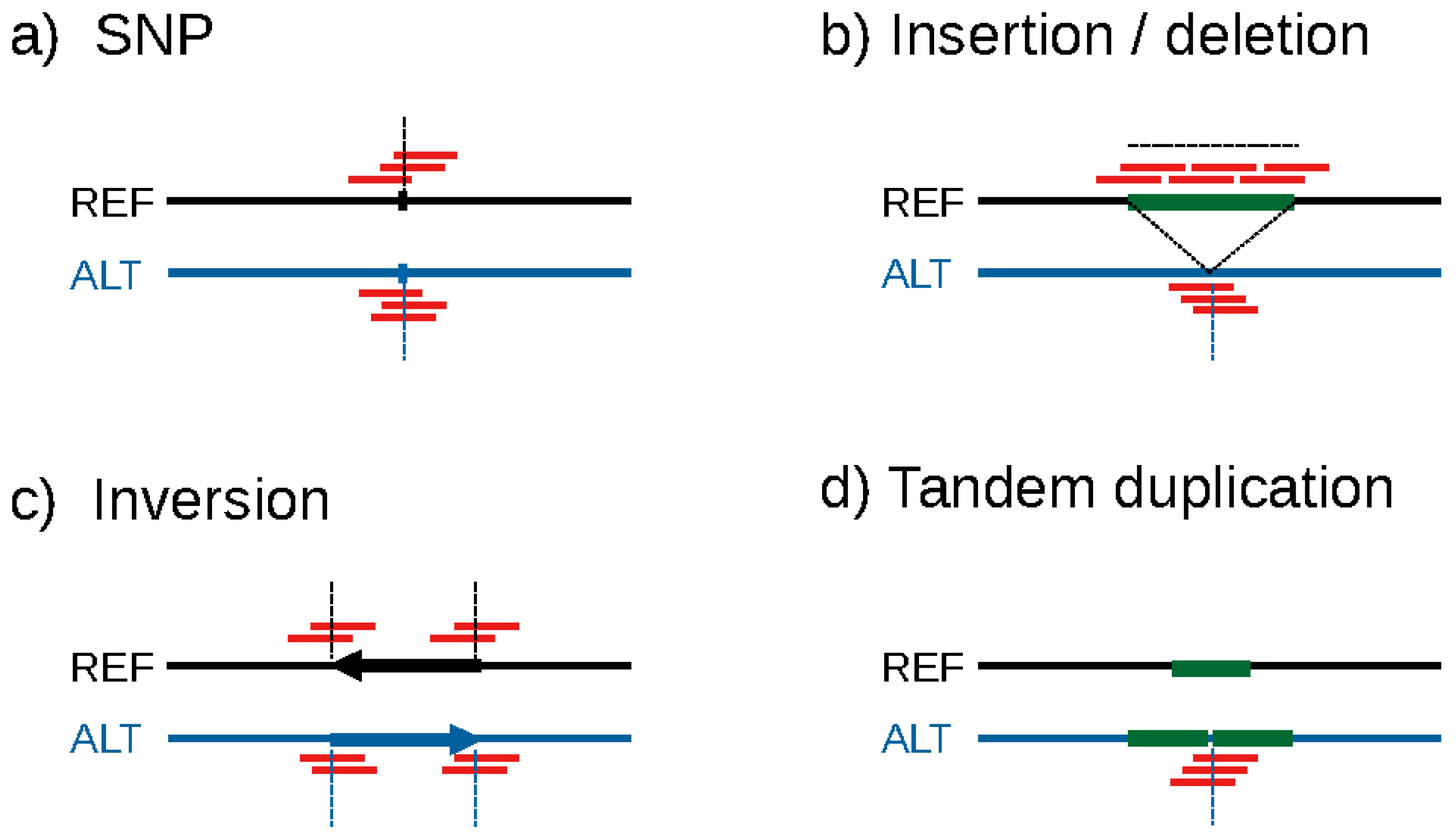
2. Limitations of Current GWAS Methods
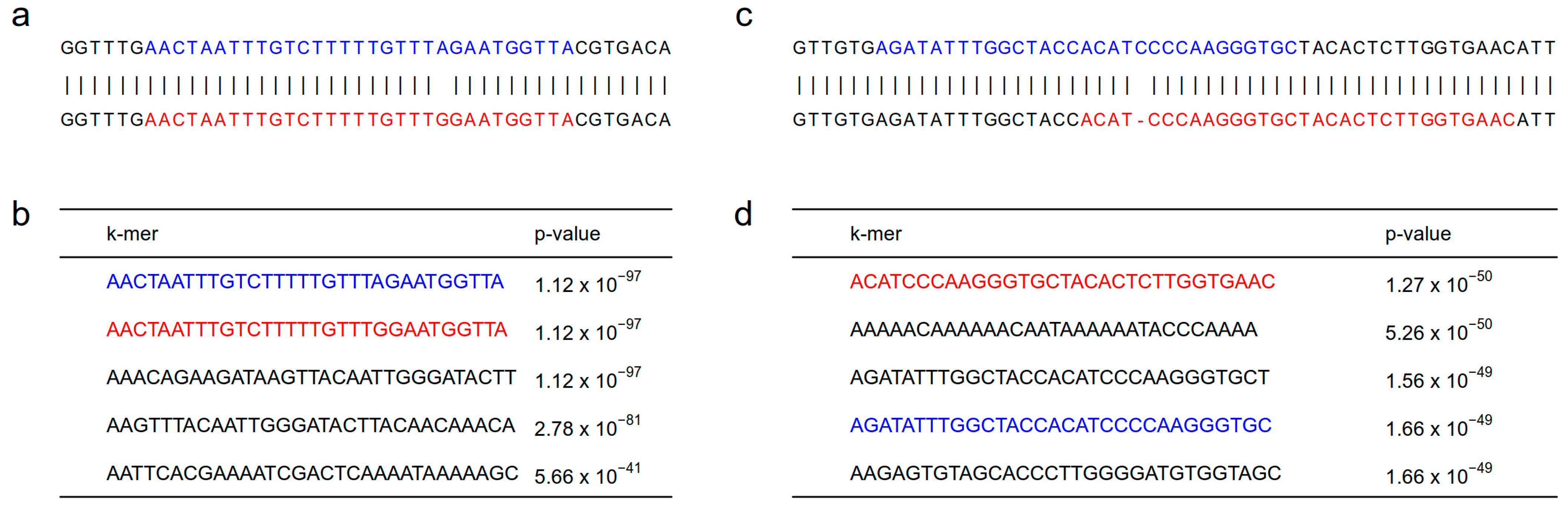
3. The Concept of k-mer-Based GWAS
4. Methods Used in k-mer-Based GWAS


5. Case Studies of k-mer-Based GWASs in Plants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crop | Number of Genotypes | Trait | Length of k-mer | Key Feature | Reference |
|---|---|---|---|---|---|
| Wild diploid wheat (Aegilops tauschii) | 195 (151 were used for phenotyping) | Stem rust (caused by Puccinia graminis f. sp. tritici) | 51 | Used sequencing data enriched for NLR genes instead of a genome-wide approach. | [22] |
| Arabidopsis | 1135 | Germination, seedling growth, flowering time, etc. | 25, 31 | Discovered new associations with structural variants and with regions missing from reference genomes. | [54] |
| Tomato | 246 | Days to tassel, ear weight, etc. | |||
| Maize | 282 | 96 metabolites, including guaiacol | |||
| Soybean | 438 Gylcine accessions | Seed pigmentation | 31 | k-mer-based approach mapped genomic region for recombinant event at I locus. | [27] |
| Maize | 282 | Upper leaf angle, flowering time, cob and kernel color, and seed oil content | 25, 31 | Used whole-genome sequencing data from the Maize 282 Association Panel (maize282) [82] to conduct both k-mer- and SNP-based GWAS. | [52] |
| Wild diploid wheat (Aegilops tauschii) | 242 | Stem rust (caused by P. graminis f. sp. tritici), powdery mildew (caused by Blumeria graminis f. sp. tritici), resistance to the wheat curl mite Aceria tosichella (vector of wheat streak mosaic virus), leaf trichomes, flowering time, and spikelet number per spike | 51 | Genome-wide extension of the method developed by Arora et al. [22]. | [59] |
| Wheat | 320, including 300 landraces | Blast fungus (caused by Pyricularia oryzae) | 51 | Functional validation of a candidate gene via virus-induced gene silencing and development of functional markers. | [73] |
| Soybean | 363 G. max | 13 traits including pod color, pubescence form, and resistance to the oomycete Phytophthora sojae | 31 | Detected several well-known loci/genes for each of the traits. | [62] |
6. Challenges and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aranzana, M.J.; Kim, S.; Zhao, K.; Bakker, E.; Horton, M.; Jakob, K.; Lister, C.; Molitor, J.; Shindo, C.; Tang, C.; et al. Genome-Wide Association Mapping in Arabidopsis Identifies Previously Known Flowering Time and Pathogen Resistance Genes. PLoS Genet. 2005, 1, e60. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, K.; Ohnishi, Y.; Iida, A.; Sekine, A.; Yamada, R.; Tsunoda, T.; Sato, H.; Sato, H.; Hori, M.; Nakamura, Y.; et al. Functional SNPs in the lymphotoxin-α gene that are associated with susceptibility to myocardial infarction. Nat. Genet. 2002, 32, 650–654. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Lander, E.S.; Hewitt, J.D.; Peterson, S.; Lincoln, S.E.; Tanksley, S.D. Resolution of quantitative traits into Mendelian factors by using a complete linkage map of restriction fragment length polymorphisms. Nature 1988, 335, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.K.; Zhang, L.; Niyitanga, S.; Afzal, M.Z.; Xu, Y.; Zhang, L.; Zhang, L.; Qi, J. Principles and approaches of association mapping in plant breeding. Trop. Plant Biol. 2020, 13, 212–224. [Google Scholar] [CrossRef]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Varshney, R.K.; Sinha, P.; Singh, V.K.; Kumar, A.; Zhang, Q.; Bennetzen, J.L. 5Gs for crop genetic improvement. Curr. Opin. Plant Biol. 2020, 56, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tamba, C.L.; Wen, Y.-J.; Li, P.; Ren, W.-L.; Ni, Y.-L.; Gao, J.; Zhang, Y.-M. mrMLM v4.0.2: An R Platform for Multi-locus Genome-wide Association Studies. Genom. Proteom. Bioinform. 2020, 18, 481–487. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.; Zhang, H.; Liu, Z.; Wang, Y.; Xing, L.; He, Q.; Du, H. Plant pan-genomics: Recent advances, new challenges, and roads ahead. J. Genet. Genom. 2022, 49, 833–846. [Google Scholar] [CrossRef]
- Sinha, P.; Singh, V.K.; Saxena, R.K.; Khan, A.W.; Abbai, R.; Chitikineni, A.; Desai, A.; Molla, J.; Upadhyaya, H.D.; Kumar, A.; et al. Superior haplotypes for haplotype-based breeding for drought tolerance in pigeonpea (Cajanus cajan L.). Plant Biotechnol. J. 2020, 18, 2482–2490. [Google Scholar] [CrossRef]
- Li, J.; Zhao, J.; Li, Y.; Gao, Y.; Hua, S.; Nadeem, M.; Sun, G.; Zhang, W.; Hou, J.; Wang, X.; et al. Identification of a novel seed size associated locus SW9-1 in soybean. Crop J. 2019, 7, 548–559. [Google Scholar] [CrossRef]
- Gupta, P.K.; Kulwal, P.L.; Jaiswal, V. Association Mapping in Crop Plants: Opportunities and Challenges. In Advances in Genetics; Friedmann, T., Dunlap, J.C., Goodwin, S.F., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 85, pp. 109–147. [Google Scholar]
- Gresham, D.; Dunham, M.J.; Botstein, D. Comparing whole genomes using DNA microarrays. Nat. Rev. Genet. 2008, 9, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J. The beginning of the end for microarrays? Nat. Methods 2008, 5, 585–587. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011, 12, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Sonah, H.; O’Donoughue, L.; Cober, E.; Rajcan, I.; Belzile, F. Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol. J. 2015, 13, 211–221. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Tibbs Cortes, L.; Zhang, Z.; Yu, J. Status and prospects of genome-wide association studies in plants. Plant Genome 2021, 14, e20077. [Google Scholar] [CrossRef]
- Abbott, J.C.; Butcher, S.A. Strategies towards sequencing complex crop genomes. Genome Biol. 2012, 13, 322. [Google Scholar] [CrossRef]
- Lu, F.; Romay, M.C.; Glaubitz, J.C.; Bradbury, P.J.; Elshire, R.J.; Wang, T.; Li, Y.; Li, Y.; Semagn, K.; Zhang, X.; et al. High-resolution genetic mapping of maize pan-genome sequence anchors. Nat. Commun. 2015, 6, 6914. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Tian, Z.; Lai, J.; Huang, X. Plant pan-genomics and its applications. Mol. Plant 2023, 16, 168–186. [Google Scholar] [CrossRef]
- Walkowiak, S.; Gao, L.; Monat, C.; Haberer, G.; Kassa, M.T.; Brinton, J.; Ramirez-Gonzalez, R.H.; Kolodziej, M.C.; Delorean, E.; Thambugala, D.; et al. Multiple wheat genomes reveal global variation in modern breeding. Nature 2020, 588, 277–283. [Google Scholar] [CrossRef]
- Arora, S.; Steuernagel, B.; Gaurav, K.; Chandramohan, S.; Long, Y.; Matny, O.; Johnson, R.; Enk, J.; Periyannan, S.; Singh, N.; et al. Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nat. Biotechnol. 2019, 37, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Zanini, S.F.; Bayer, P.E.; Wells, R.; Snowdon, R.J.; Batley, J.; Varshney, R.K.; Nguyen, H.T.; Edwards, D.; Golicz, A.A. Pangenomics in crop improvement—From coding structural variations to finding regulatory variants with pangenome graphs. Plant Genome 2022, 15, e20177. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K. GWAS for genetics of complex quantitative traits: Genome to pangenome and SNPs to SVs and k-mers. BioEssays 2021, 43, 2100109. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K. Quantitative genetics: Pan-genomes, SVs, and k-mers for GWAS. Trends Genet. 2021, 37, 868–871. [Google Scholar] [CrossRef]
- Jin, S.; Han, Z.; Hu, Y.; Si, Z.; Dai, F.; He, L.; Cheng, Y.; Li, Y.; Zhao, T.; Fang, L.; et al. Structural variation (SV)-based pan-genome and GWAS reveal the impacts of SVs on the speciation and diversification of allotetraploid cottons. Mol. Plant 2023, 16, 678–693. [Google Scholar] [CrossRef]
- Kim, J.-H.; Park, J.-S.; Lee, C.-Y.; Jeong, M.-G.; Xu, J.L.; Choi, Y.; Jung, H.-W.; Choi, H.-K. Dissecting seed pigmentation-associated genomic loci and genes by employing dual approaches of reference-based and k-mer-based GWAS with 438 Glycine accessions. PLoS ONE 2020, 15, e0243085. [Google Scholar] [CrossRef]
- Alonge, M.; Wang, X.; Benoit, M.; Soyk, S.; Pereira, L.; Zhang, L.; Suresh, H.; Ramakrishnan, S.; Maumus, F.; Ciren, D.; et al. Major Impacts of Widespread Structural Variation on Gene Expression and Crop Improvement in Tomato. Cell 2020, 182, 145–161.e123. [Google Scholar] [CrossRef]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Hsi-Yang Fritz, M.; et al. An integrated map of structural variation in 2504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [Green Version]
- De Coster, W.; Van Broeckhoven, C. Newest Methods for Detecting Structural Variations. Trends Biotechnol. 2019, 37, 973–982. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.S.; Urban, A.E.; Mills, R.E. Structural variation in the sequencing era. Nat. Rev. Genet. 2020, 21, 171–189. [Google Scholar] [CrossRef]
- Bridges, C.B. The Bar “gene” a duplication. Science 1936, 83, 210–211. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.E.; Lee, T.G.; Guo, X.; Melito, S.; Wang, K.; Bayless, A.M.; Wang, J.; Hughes, T.J.; Willis, D.K.; Clemente, T.E.; et al. Copy Number Variation of Multiple Genes at Rhg1 Mediates Nematode Resistance in Soybean. Science 2012, 338, 1206–1209. [Google Scholar] [CrossRef] [Green Version]
- Gabur, I.; Chawla, H.S.; Snowdon, R.J.; Parkin, I.A.P. Connecting genome structural variation with complex traits in crop plants. Theor. Appl. Genet. 2019, 132, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Zhang, X.; Zhang, S.; Lin, Z.; Zhang, X.; Ming, R. Structural variations in papaya genomes. BMC Genom. 2021, 22, 335. [Google Scholar] [CrossRef]
- Maron, L.G.; Guimarães, C.T.; Kirst, M.; Albert, P.S.; Birchler, J.A.; Bradbury, P.J.; Buckler, E.S.; Coluccio, A.E.; Danilova, T.V.; Kudrna, D.; et al. Aluminum tolerance in maize is associated with higher MATE1 gene copy number. Proc. Natl. Acad. Sci. USA 2013, 110, 5241–5246. [Google Scholar] [CrossRef]
- Song, J.-M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Sun, M.; Zhang, Z.; Jin, Y.; Zhang, A.; Lin, C.; Wu, B.; He, M.; Xu, B.; Wang, J.; et al. Pangenomic analysis identifies structural variation associated with heat tolerance in pearl millet. Nat. Genet. 2023, 55, 507–518. [Google Scholar] [CrossRef]
- Studer, A.; Zhao, Q.; Ross-Ibarra, J.; Doebley, J. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 2011, 43, 1160–1163. [Google Scholar] [CrossRef] [Green Version]
- Jayakodi, M.; Schreiber, M.; Stein, N.; Mascher, M. Building pan-genome infrastructures for crop plants and their use in association genetics. DNA Res. 2021, 28, dsaa030. [Google Scholar] [CrossRef]
- Liu, Y.; Du, H.; Li, P.; Shen, Y.; Peng, H.; Liu, S.; Zhou, G.-A.; Zhang, H.; Liu, Z.; Shi, M.; et al. Pan-Genome of Wild and Cultivated Soybeans. Cell 2020, 182, 162–176.e113. [Google Scholar] [CrossRef]
- Li, H.; Wang, S.; Chai, S.; Yang, Z.; Zhang, Q.; Xin, H.; Xu, Y.; Lin, S.; Chen, X.; Yao, Z.; et al. Graph-based pan-genome reveals structural and sequence variations related to agronomic traits and domestication in cucumber. Nat. Commun. 2022, 13, 682. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, M.; Dugas, E.; Benchouaia, M.; Leduque, B.; Jiménez-Gómez, J.M.; Colot, V.; Quadrana, L. The impact of transposable elements on tomato diversity. Nat. Commun. 2020, 11, 4058. [Google Scholar] [CrossRef]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Willson, J. Resolving the roles of structural variants. Nat. Rev. Genet. 2020, 21, 507. [Google Scholar] [CrossRef] [PubMed]
- Della Coletta, R.; Qiu, Y.; Ou, S.; Hufford, M.B.; Hirsch, C.N. How the pan-genome is changing crop genomics and improvement. Genome Biol. 2021, 22, 3. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Gonda, I.; Sun, H.; Ma, Q.; Bao, K.; Tieman, D.M.; Burzynski-Chang, E.A.; Fish, T.L.; Stromberg, K.A.; Sacks, G.L.; et al. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 2019, 51, 1044–1051. [Google Scholar] [CrossRef]
- Li, C.; Xiang, X.; Huang, Y.; Zhou, Y.; An, D.; Dong, J.; Zhao, C.; Liu, H.; Li, Y.; Wang, Q.; et al. Long-read sequencing reveals genomic structural variations that underlie creation of quality protein maize. Nat. Commun. 2020, 11, 17. [Google Scholar] [CrossRef] [Green Version]
- Pucker, B.; Irisarri, I.; de Vries, J.; Xu, B. Plant genome sequence assembly in the era of long reads: Progress, challenges and future directions. Quant. Plant Biol. 2022, 3, e5. [Google Scholar] [CrossRef]
- Zhang, F.; Xue, H.; Dong, X.; Li, M.; Zheng, X.; Li, Z.; Xu, J.; Wang, W.; Wei, C. Long-read sequencing of 111 rice genomes reveals significantly larger pan-genomes. Genome Res. 2022, 32, 853–863. [Google Scholar] [CrossRef]
- Torkamaneh, D.; Laroche, J.; Tardivel, A.; O’Donoughue, L.; Cober, E.; Rajcan, I.; Belzile, F. Comprehensive description of genomewide nucleotide and structural variation in short-season soybean. Plant Biotechnol. J. 2017, 16, 749–759. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Washburn, J.D.; Hao, Y.; Zhang, Z.; Yang, J.; Liu, S. Trait Association and Prediction Through Integrative K-mer Analysis. bioRxiv 2021, 2021-11. [Google Scholar] [CrossRef]
- Rahman, A.; Hallgrímsdóttir, I.; Eisen, M.; Pachter, L. Association mapping from sequencing reads using k-mers. eLife 2018, 7, e32920. [Google Scholar] [CrossRef] [PubMed]
- Voichek, Y.; Weigel, D. Identifying genetic variants underlying phenotypic variation in plants without complete genomes. Nat. Genet. 2020, 52, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 2047-217X. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.-D.; Liu, X.-M.; Huang, T.-L.; Xia, L.-C. The statistical power of k-mer based aggregative statistics for alignment-free detection of horizontal gene transfer. Synth. Syst. Biotechnol. 2019, 4, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Ebler, J.; Ebert, P.; Clarke, W.E.; Rausch, T.; Audano, P.A.; Houwaart, T.; Mao, Y.; Korbel, J.O.; Eichler, E.E.; Zody, M.C.; et al. Pangenome-based genome inference allows efficient and accurate genotyping across a wide spectrum of variant classes. Nat. Genet. 2022, 54, 518–525. [Google Scholar] [CrossRef]
- Sibbesen, J.A.; Maretty, L.; Krogh, A.; The Danish Pan-Genome, C. Accurate genotyping across variant classes and lengths using variant graphs. Nat. Genet. 2018, 50, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Gaurav, K.; Arora, S.; Silva, P.; Sánchez-Martín, J.; Horsnell, R.; Gao, L.; Brar, G.S.; Widrig, V.; John Raupp, W.; Singh, N.; et al. Population genomic analysis of Aegilops tauschii identifies targets for bread wheat improvement. Nat. Biotechnol. 2022, 40, 422–431. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Q.; Deng, C.; Zheng, Y.; Sun, F. KmerGO: A tool to identify group-specific sequences with k-mers. Front. Microbiol. 2020, 11, 2067. [Google Scholar] [CrossRef]
- Koren, S.; Rhie, A.; Walenz, B.P.; Dilthey, A.T.; Bickhart, D.M.; Kingan, S.B.; Hiendleder, S.; Williams, J.L.; Smith, T.P.L.; Phillippy, A.M. De novo assembly of haplotype-resolved genomes with trio binning. Nat. Biotechnol. 2018, 36, 1174–1182. [Google Scholar] [CrossRef]
- Lemay, M.-A.; de Ronne, M.; Bélanger, R.; Belzile, F. k-mer-based GWAS enhances the discovery of causal variants and candidate genes in soybean. bioRxiv 2023, 2023-03. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhang, Z. GAPIT Version 3: Boosting Power and Accuracy for Genomic Association and Prediction. Genom. Proteom. Bioinform. 2021, 19, 629–640. [Google Scholar] [CrossRef]
- Kokot, M.; Długosz, M.; Deorowicz, S. KMC 3: Counting and manipulating k-mer statistics. Bioinformatics 2017, 33, 2759–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manekar, S.C.; Sathe, S.R. A benchmark study of k-mer counting methods for high-throughput sequencing. GigaScience 2018, 7, giy125. [Google Scholar] [CrossRef] [Green Version]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhie, A.; Walenz, B.P.; Koren, S.; Phillippy, A.M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020, 21, 245. [Google Scholar] [CrossRef]
- Melsted, P.; Pritchard, J.K. Efficient counting of k-mers in DNA sequences using a bloom filter. BMC Bioinform. 2011, 12, 333. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [Green Version]
- Lemane, T.; Chikhi, R.; Peterlongo, P. kmdiff, large-scale and user-friendly differential k-mer analyses. Bioinformatics 2022, 38, 5443–5445. [Google Scholar] [CrossRef]
- Wang, M.; Li, W.; Fang, C.; Xu, F.; Liu, Y.; Wang, Z.; Yang, R.; Zhang, M.; Liu, S.; Lu, S.; et al. Parallel selection on a dormancy gene during domestication of crops from multiple families. Nat. Genet. 2018, 50, 1435–1441. [Google Scholar] [CrossRef]
- Arora, S.; Steed, A.; Goddard, R.; Gaurav, K.; O’Hara, T.; Schoen, A.; Rawat, N.; Elkot, A.F.; Korolev, A.V.; Chinoy, C.; et al. A wheat kinase and immune receptor form host-specificity barriers against the blast fungus. Nat. Plants 2023, 9, 385–392. [Google Scholar] [CrossRef]
- Allen, J.P.; Snitkin, E.; Pincus, N.B.; Hauser, A.R. Forest and Trees: Exploring Bacterial Virulence with Genome-wide Association Studies and Machine Learning. Trends Microbiol. 2021, 29, 621–633. [Google Scholar] [CrossRef]
- Aun, E.; Brauer, A.; Kisand, V.; Tenson, T.; Remm, M. A k-mer-based method for the identification of phenotype-associated genomic biomarkers and predicting phenotypes of sequenced bacteria. PLoS Comput. Biol. 2018, 14, e1006434. [Google Scholar] [CrossRef] [Green Version]
- Jaillard, M.; Lima, L.; Tournoud, M.; Mahé, P.; van Belkum, A.; Lacroix, V.; Jacob, L. A fast and agnostic method for bacterial genome-wide association studies: Bridging the gap between k-mers and genetic events. PLoS Genet. 2018, 14, e1007758. [Google Scholar] [CrossRef] [Green Version]
- Jaillard, M.; Palmieri, M.; van Belkum, A.; Mahé, P. Interpreting k-mer–based signatures for antibiotic resistance prediction. GigaScience 2020, 9, giaa110. [Google Scholar] [CrossRef]
- Yu, G.; Zhang, Q.; Friesen, T.L.; Rouse, M.N.; Jin, Y.; Zhong, S.; Rasmussen, J.B.; Lagudah, E.S.; Xu, S.S. Identification and mapping of Sr46 from Aegilops tauschii accession CIae 25 conferring resistance to race TTKSK (Ug99) of wheat stem rust pathogen. Theor. Appl. Genet. 2015, 128, 431–443. [Google Scholar] [CrossRef]
- Olson, E.L.; Rouse, M.N.; Pumphrey, M.O.; Bowden, R.L.; Gill, B.S.; Poland, J.A. Simultaneous transfer, introgression, and genomic localization of genes for resistance to stem rust race TTKSK (Ug99) from Aegilops tauschii to wheat. Theor. Appl. Genet. 2013, 126, 1179–1188. [Google Scholar] [CrossRef]
- Rouse, M.N.; Olson, E.L.; Gill, B.S.; Pumphrey, M.O.; Jin, Y. Stem Rust Resistance in Aegilops tauschii Germplasm. Crop Sci. 2011, 51, 2074–2078. [Google Scholar] [CrossRef]
- Steuernagel, B.; Periyannan, S.K.; Hernández-Pinzón, I.; Witek, K.; Rouse, M.N.; Yu, G.; Hatta, A.; Ayliffe, M.; Bariana, H.; Jones, J.D.G.; et al. Rapid cloning of disease-resistance genes in plants using mutagenesis and sequence capture. Nat. Biotechnol. 2016, 34, 652–655. [Google Scholar] [CrossRef]
- Flint-Garcia, S.A.; Thuillet, A.-C.; Yu, J.; Pressoir, G.; Romero, S.M.; Mitchell, S.E.; Doebley, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Maize association population: A high-resolution platform for quantitative trait locus dissection. Plant J. 2005, 44, 1054–1064. [Google Scholar] [CrossRef]
- Yan, L.; Fu, D.; Li, C.; Blechl, A.; Tranquilli, G.; Bonafede, M.; Sanchez, A.; Valarik, M.; Yasuda, S.; Dubcovsky, J. The wheat and barley vernalization gene VRN3 is an orthologue of FT. Proc. Natl. Acad. Sci. USA 2006, 103, 19581–19586. [Google Scholar] [CrossRef]
- Bonnin, I.; Rousset, M.; Madur, D.; Sourdille, P.; Dupuits, C.; Brunel, D.; Goldringer, I. FT genome A and D polymorphisms are associated with the variation of earliness components in hexaploid wheat. Theor. Appl. Genet. 2008, 116, 383–394. [Google Scholar] [CrossRef]
- Dixon, L.E.; Farré, A.; Finnegan, E.J.; Orford, S.; Griffiths, S.; Boden, S.A. Developmental responses of bread wheat to changes in ambient temperature following deletion of a locus that includes FLOWERING LOCUS T1. Plant Cell Environ. 2018, 41, 1715–1725. [Google Scholar] [CrossRef] [Green Version]
- Claeys, H.; Vi, S.L.; Xu, X.; Satoh-Nagasawa, N.; Eveland, A.L.; Goldshmidt, A.; Feil, R.; Beggs, G.A.; Sakai, H.; Brennan, R.G.; et al. Control of meristem determinacy by trehalose 6-phosphate phosphatases is uncoupled from enzymatic activity. Nat. Plants 2019, 5, 352–357. [Google Scholar] [CrossRef]
- Koppolu, R.; Anwar, N.; Sakuma, S.; Tagiri, A.; Lundqvist, U.; Pourkheirandish, M.; Rutten, T.; Seiler, C.; Himmelbach, A.; Ariyadasa, R.; et al. Six-rowed spike4 (Vrs4) controls spikelet determinacy and row-type in barley. Proc. Natl. Acad. Sci. USA 2013, 110, 13198–13203. [Google Scholar] [CrossRef]
- Malik, R.; Brown-Guedira, G.L.; Smith, C.M.; Harvey, T.L.; Gill, B.S. Genetic Mapping of Wheat Curl Mite Resistance Genes Cmc3 and Cmc4 in Common Wheat. Crop Sci. 2003, 43, 644–650. [Google Scholar] [CrossRef]
- Zhao, J.; Abdelsalam, N.R.; Khalaf, L.; Chuang, W.-P.; Zhao, L.; Smith, C.M.; Carver, B.; Bai, G. Development of Single Nucleotide Polymorphism Markers for the Wheat Curl Mite Resistance Gene Cmc4. Crop Sci. 2019, 59, 1567–1575. [Google Scholar] [CrossRef]
- Klymiuk, V.; Yaniv, E.; Huang, L.; Raats, D.; Fatiukha, A.; Chen, S.; Feng, L.; Frenkel, Z.; Krugman, T.; Lidzbarsky, G.; et al. Cloning of the wheat Yr15 resistance gene sheds light on the plant tandem kinase-pseudokinase family. Nat. Commun. 2018, 9, 3735. [Google Scholar] [CrossRef] [Green Version]
- Brueggeman, R.; Rostoks, N.; Kudrna, D.; Kilian, A.; Han, F.; Chen, J.; Druka, A.; Steffenson, B.; Kleinhofs, A. The barley stem rust-resistance gene Rpg1 is a novel disease-resistance gene with homology to receptor kinases. Proc. Natl. Acad. Sci. USA 2002, 99, 9328–9333. [Google Scholar] [CrossRef]
- Chen, S.; Rouse, M.N.; Zhang, W.; Zhang, X.; Guo, Y.; Briggs, J.; Dubcovsky, J. Wheat gene Sr60 encodes a protein with two putative kinase domains that confers resistance to stem rust. New Phytol. 2020, 225, 948–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Guo, L.; Wang, Z.; Li, B.; Li, J.; Li, Y.; Qiu, D.; Shi, W.; Yang, L.; Wang, N.; et al. A rare gain of function mutation in a wheat tandem kinase confers resistance to powdery mildew. Nat. Commun. 2020, 11, 680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, Y.; Vy, T.T.P.; Yoshida, K.; Asano, H.; Mitsuoka, C.; Asuke, S.; Anh, V.L.; Cumagun, C.J.R.; Chuma, I.; Terauchi, R.; et al. Evolution of the wheat blast fungus through functional losses in a host specificity determinant. Science 2017, 357, 80–83. [Google Scholar] [CrossRef] [Green Version]
- Vy, T.T.P.; Hyon, G.-S.; Nga, N.T.T.; Inoue, Y.; Chuma, I.; Tosa, Y. Genetic analysis of host–pathogen incompatibility between Lolium isolates of Pyricularia oryzae and wheat. J. Gen. Plant Pathol. 2014, 80, 59–65. [Google Scholar] [CrossRef]
- Hirata, K.; Tosa, Y.; Nakayashiki, H.; Mayama, S. Significance of PWT4–Rwt4 interaction in the species specificity of Avena isolates of Magnaporthe oryzae on wheat. J. Gen. Plant Pathol. 2005, 71, 340–344. [Google Scholar] [CrossRef]
- Rawat, N.; Joshi, A.; Pumphrey, M.; Singh, L.; Mahlandt, A.; Chhabra, B.; Wilson, D.; Gill, B.; Poland, J.; Tiwari, V. A TILLING Resource for Hard Red Winter Wheat Variety Jagger. Crop Sci. 2019, 59, 1666–1671. [Google Scholar] [CrossRef]
- Zabala, G.; Vodkin, L.O. A Rearrangement Resulting in Small Tandem Repeats in the F3′5′H Gene of White Flower Genotypes Is Associated with the Soybean W1 Locus. Crop Sci. 2007, 47, S-113–S-124. [Google Scholar] [CrossRef]
- Zabala, G.; Vodkin, L. Cloning of the pleiotropic T locus in soybean and two recessive alleles that differentially affect structure and expression of the encoded flavonoid 3’ hydroxylase. Genetics 2003, 163, 295–309. [Google Scholar] [CrossRef]
- Yan, F.; Githiri, S.M.; Liu, Y.; Sang, Y.; Wang, Q.; Takahashi, R. Loss-of-Function Mutation of Soybean R2R3 MYB Transcription Factor Dilutes Tawny Pubescence Color. Front. Plant Sci. 2020, 10, 1809. [Google Scholar] [CrossRef] [Green Version]
- Corut, K. akcorut/kGWASflow: V1.2.0 (v1.2.0). Zenodo 2023. [Google Scholar] [CrossRef]
- Eizenga, J.M.; Novak, A.M.; Sibbesen, J.A.; Heumos, S.; Ghaffaari, A.; Hickey, G.; Chang, X.; Seaman, J.D.; Rounthwaite, R.; Ebler, J.; et al. Pangenome Graphs. Annu. Rev. Genom. Hum. Genet. 2020, 21, 139–162. [Google Scholar] [CrossRef] [PubMed]
- Garrison, E.; Sirén, J.; Novak, A.M.; Hickey, G.; Eizenga, J.M.; Dawson, E.T.; Jones, W.; Garg, S.; Markello, C.; Lin, M.F.; et al. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 2018, 36, 875–879. [Google Scholar] [CrossRef]
- Hickey, G.; Monlong, J.; Ebler, J.; Novak, A.M.; Eizenga, J.M.; Gao, Y.; Abel, H.J.; Antonacci-Fulton, L.L.; Asri, M.; Baid, G.; et al. Pangenome graph construction from genome alignments with Minigraph-Cactus. Nat. Biotechnol. 2023, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Zhang, H.; Jain, C.; Aluru, S. A comprehensive evaluation of long read error correction methods. BMC Genom. 2020, 21, 889. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karikari, B.; Lemay, M.-A.; Belzile, F. k-mer-Based Genome-Wide Association Studies in Plants: Advances, Challenges, and Perspectives. Genes 2023, 14, 1439. https://doi.org/10.3390/genes14071439
Karikari B, Lemay M-A, Belzile F. k-mer-Based Genome-Wide Association Studies in Plants: Advances, Challenges, and Perspectives. Genes. 2023; 14(7):1439. https://doi.org/10.3390/genes14071439
Chicago/Turabian StyleKarikari, Benjamin, Marc-André Lemay, and François Belzile. 2023. "k-mer-Based Genome-Wide Association Studies in Plants: Advances, Challenges, and Perspectives" Genes 14, no. 7: 1439. https://doi.org/10.3390/genes14071439