
Arabidopsis Transcription Regulatory Factor Domain/Domain Interaction Analysis Tool—Liquid/Liquid Phase Separation, Oligomerization, GO Analysis: A Toolkit for Interaction Data-Based Domain Analysis

 ,
,
Abstract
:1. Introduction
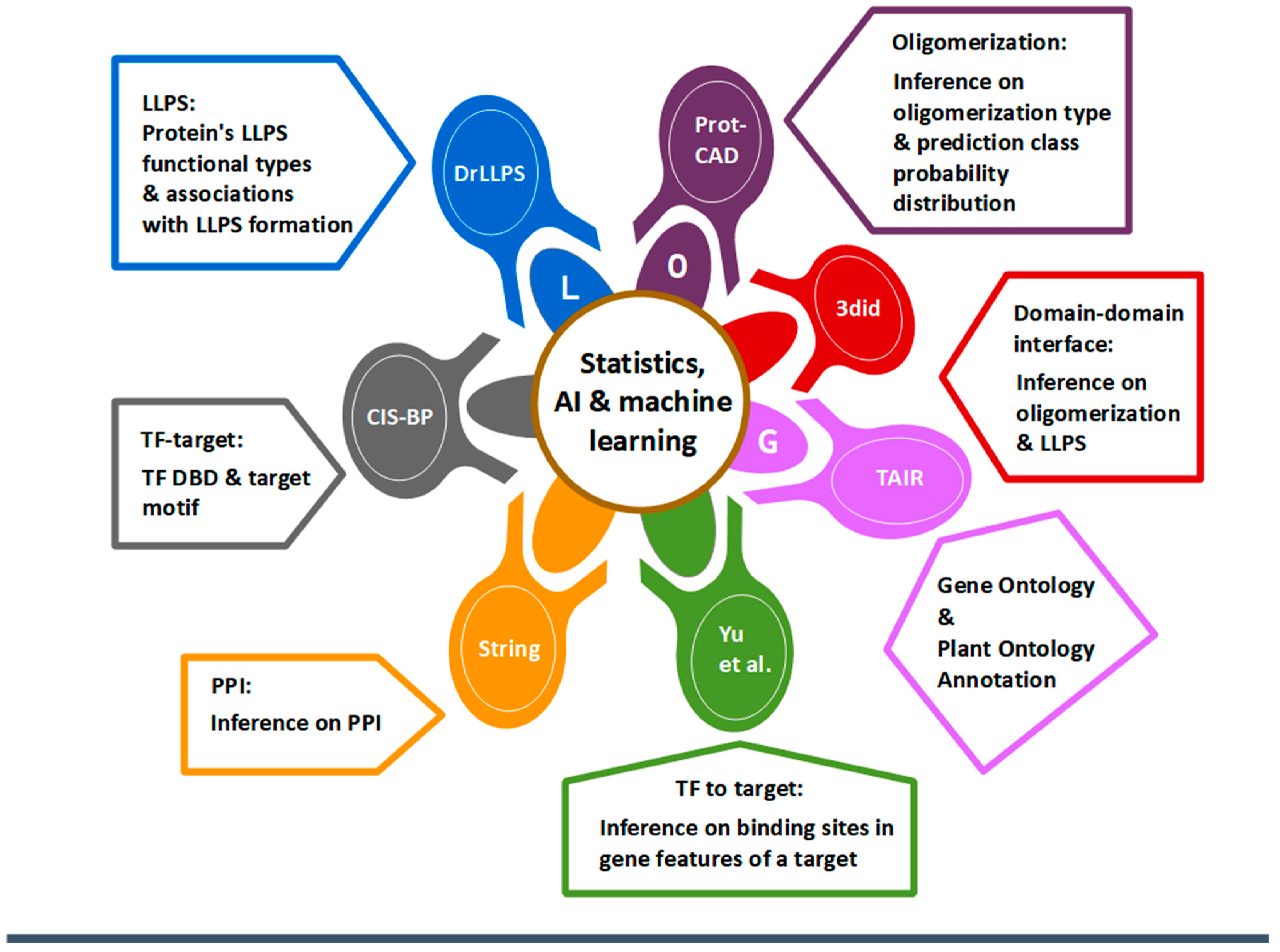
2. Methods
2.1. Oligomerization Module
2.2. DDI Module
2.3. LLPS Module
2.4. GO Analysis Module
2.5. TF-Target Module
2.6. PPI Module
2.7. TF-to-Target Module
2.8. Proof of Concept of Search Algorithm
2.9. Demonstration of Program Usage
2.9.1. Prediction of Oligomerization Types and LLPS Types
2.9.2. Prediction of Oligomerization Types and Correlation Analysis
2.9.3. Prediction of TF Binding Motif Types and PPI/PNI Study
3. Results
3.1. Prediction of Oligomerization Types and LLPS Types
3.2. Prediction and Extraction of Important Features from TF-LLPS Factor Data
3.3. Prediction of TF Binding Motif Types and PPI/PNI Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verde, I.; The International Peach Genome Initiative; Abbott, A.G.; Scalabrin, S.; Jung, S.; Shu, S.; Marroni, F.; Zhebentyayeva, T.; Dettori, M.T.; Grimwood, J.; et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013, 45, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Cao, K.; Zhou, Z.; Wang, Q.; Guo, J.; Zhao, P.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Wang, X.; et al. Genome-wide association study of 12 agronomic traits in peach. Nat. Commun. 2016, 7, 13246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiers, M.W.E.J.; Minnoye, L.; Aibar, S.; González-Blas, C.B.; Atak, Z.K.; Aerts, S. Mapping gene regulatory networks from single-cell omics data. Briefings Funct. Genom. 2018, 17, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Q.; Beuchat, G.; Zeng, H.; Zhang, C.; Chen, L.Q. Combined analyses of translatome and transcriptome in Arabidopsis reveal new players responding to magnesium deficiency. J. Integr. Plant Biol. 2021, 63, 2075–2092. [Google Scholar] [CrossRef]
- Mosca, R.; Céol, A.; Stein, A.; Olivella, R.; Aloy, P. 3did: A catalog of domain-based interactions of known three-dimensional structure. Nucleic Acids Res. 2013, 42, D374–D379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, X.; Zhao, F.; Wang, Y.; Liu, J.; Zhuang, Y.; Ye, L.; Qi, M.; Cheng, J.; Zhang, Y. Plant Regulomics: A data-driven interface for retrieving upstream regulators from plant multi-omics data. Plant J. 2020, 101, 237–248. [Google Scholar] [CrossRef]
- Shim, S.; Park, C.-M.; Seo, P.J. iRegNet: An integrative Regulatory Network analysis tool for Arabidopsis thaliana. Plant Physiol. 2021, 187, 1292–1309. [Google Scholar] [CrossRef]
- Su, Q.; Mehta, S.; Zhang, J. Liquid-liquid phase separation: Orchestrating cell signaling through time and space. Mol. Cell 2021, 81, 4137–4146. [Google Scholar] [CrossRef]
- Peng, P.-H.; Hsu, K.-W.; Wu, K.-J. Liquid-liquid phase separation (LLPS) in cellular physiology and tumor biology. Am. J. Cancer Res. 2021, 11, 3766–3776. [Google Scholar]
- Zhao, Y.G.; Zhang, H. Phase Separation in Membrane Biology: The Interplay between Membrane-Bound Organelles and Membraneless Condensates. Dev. Cell 2020, 55, 30–44. [Google Scholar] [CrossRef]
- Nesterov, S.V.; Ilyinsky, N.S.; Uversky, V.N. Liquid-liquid phase separation as a common organizing principle of intracellular space and biomembranes providing dynamic adaptive responses. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119102. [Google Scholar] [CrossRef]
- Li, J.; Zhang, M.; Ma, W.; Yang, B.; Lu, H.; Zhou, F.; Zhang, L. Post-translational modifications in liquid-liquid phase separation: A comprehensive review. Mol. Biomed. 2022, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Stoyle, C.L.; Stephens, P.E.; Humphreys, D.P.; Heywood, S.; Cain, K.; Bulleid, N.J. IgG light chain-independent secretion of heavy chain dimers: Consequence for therapeutic antibody production and design. Biochem. J. 2017, 474, 3179–3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, W.; Cheng, S.; Li, Y.; Li, X.-Y.; Lu, N.; Sun, J.; Tang, G.; Yang, Y.; Cai, K.; Li, X.; et al. Phase separation modulates the assembly and dynamics of a polarity-related scaffold-signaling hub. Nat. Commun. 2022, 13, 7181. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.W.; Swift, S.; Lord, C.J.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009, 10, 990–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehler Lydia, C.; Grese Zachary, R.; Bastos Alliny, C.S.; Mamede Lohany, D.; Heyduk Tomasz Ayala Yuna, M. TDP-43 Oli-gomerization and Phase Separation Properties Are Necessary for Autoregulation. Front. Neurosci. 2022, 16, 818655. [Google Scholar] [CrossRef]
- Stein, A.; Aloy, P. Novel Peptide-Mediated Interactions Derived from High-Resolution 3-Dimensional Structures. PLoS Comput. Biol. 2010, 6, e1000789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Perumal, N.B.; Oldfield, C.J.; Su, E.W.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in transcription factors. Biochemistry 2006, 45, 6873–6888. [Google Scholar] [CrossRef] [Green Version]
- Puranik, S.; Acajjaoui, S.; Conn, S.; Costa, L.; Conn, V.; Vial, A.; Marcellin, R.; Melzer, R.; Brown, E.; Hart, D.; et al. Structural Basis for the Oligomerization of the MADS Domain Transcription Factor SEPALLATA3 in Arabidopsis. Plant Cell 2014, 26, 3603–3615. [Google Scholar] [CrossRef] [Green Version]
- Sayou, C.; Nanao, M.H.; Jamin, M.; Posé, D.; Thévenon, E.; Grégoire, L.; Tichtinsky, G.; Denay, G.; Ott, F.; Llobet, M.P.; et al. A SAM oligomerization domain shapes the genomic binding landscape of the LEAFY transcription factor. Nat. Commun. 2016, 7, 11222. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Hata, N.; Banerjee, N.; Futcher, B.; Zhang, M.Q. Identifying combinatorial regulation of transcription factors and binding motifs. Genome Biol. 2004, 5, R56. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Burgos, I.; Espinosa, J.R.; Joseph, J.A.; Collepardo-Guevara, R. RNA length has a non-trivial effect in the stability of bio-molecular condensates formed by RNA-binding proteins. PLoS Comput. Biol. 2022, 18, e1009810. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Dunbrack, R.L., Jr. The protein common assembly database (ProtCAD)—A comprehensive structural resource of protein complexes. Nucleic Acids Res. 2022, 51, D466–D478. [Google Scholar] [CrossRef] [PubMed]
- Kurotani, A.; Yamada, Y.; Shinozaki, K.; Kuroda, Y.; Sakurai, T. Plant-PrAS: A Database of Physicochemical and Structural Properties and Novel Functional Regions in Plant Proteomes. Plant Cell Physiol. 2015, 56, e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, H.; Zhang, Q.; Wang, P.; Cao, B.; Jia, C.; Cheng, B.; Shi, Y.; Guo, W.-F.; Wang, Z.; Liu, Z.-X.; et al. qPTMplants: An integrative database of quantitative post-translational modifications in plants. Nucleic Acids Res. 2022, 50, D1491–D1499. [Google Scholar] [CrossRef] [PubMed]
- Ning, W.; Guo, Y.; Lin, S.; Mei, B.; Wu, Y.; Jiang, P.; Tan, X.; Zhang, W.; Chen, G.; Peng, D.; et al. DrLLPS: A data resource of liquid–liquid phase separation in eukaryotes. Nucleic Acids Res. 2020, 48, D288–D295. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Beavis, W.; Berardini, T.Z.; Chen, G.; Dixon, D.; Doyle, A.; Garcia-Hernandez, M.; Huala, E.; Lander, G.; Montoya, M.; et al. The Arabidopsis Information Resource (TAIR): A model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res. 2003, 31, 224–228. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Walls, R.L.; Cooper, L.; Elser, J.; Gandolfo, M.A.; Mungall, C.J.; Smith, B.; Stevenson, D.W.; Jaiswal, P. The Plant Ontology Facilitates Comparisons of Plant Development Stages Across Species. Front. Plant Sci. 2019, 10, 631. [Google Scholar] [CrossRef]
- Weirauch, M.T.; Yang, A.; Albu, M.; Cote, A.G.; Montenegro-Montero, A.; Drewe, P.; Najafabadi, H.S.; Lambert, S.A.; Mann, I.; Cook, K.; et al. Determination and Inference of Eukaryotic Transcription Factor Sequence Specificity. Cell 2014, 158, 1431–1443. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujato, M.; Kieken, F.; Skiles, A.A.; Tapinos, N.; Fiser, A. Prediction of DNA binding motifs from 3D models of transcription factors; identifying TLX3 regulated genes. Nucleic Acids Res. 2014, 42, 13500–13512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.-P.; Lin, J.-J.; Li, W.-H. Positional distribution of transcription factor binding sites in Arabidopsis thaliana. Sci. Rep. 2016, 6, 25164. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, Q.; Huang, H.; Huang, W.; Chen, Y.; McGarvey, P.B.; Wu, C.H.; Arighi, C.N.; on behalf of the UniProt Consortium. A crowdsourcing open platform for literature curation in UniProt. PLoS Biol. 2021, 19, e3001464. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res. 2022, 51, D418–D427. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 2015, 53, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Degtyareva, A.O.; Antontseva, E.V.; Merkulova, T.I. Regulatory SNPs: Altered Transcription Factor Binding Sites Implicated in Complex Traits and Diseases. Int. J. Mol. Sci. 2021, 22, 6454. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Goncearenco, A.; Berezovsky, I.N. Nucleotide binding database NBDB--a collection of sequence motifs with specific protein-ligand interactions. Nucleic Acids Res. 2016, 44, D301–D307. [Google Scholar] [CrossRef] [Green Version]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztányi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D2P2: Database of disordered protein predictions. Nucleic Acids Res. 2013, 41, D508–D516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, U.; Schmeier, S.; Bajic, V.B. TcoF-DB: Dragon database for human transcription co-factors and transcription factor interacting proteins. Nucleic Acids Res. 2011, 39, D106–D110. [Google Scholar] [CrossRef]
- Palaniswamy, S.K.; James, S.; Sun, H.; Lamb, R.S.; Davuluri, R.V.; Grotewold, E. AGRIS and AtRegNet. A Platform to Link cis-Regulatory Elements and Transcription Factors into Regulatory Networks. Plant Physiol. 2006, 140, 818–829. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wei, L.; Wang, C.; Zhao, J.; Han, S.; Zhang, Y.; Du, W. LPInsider: A webserver for lncRNA–protein interaction extraction from the literature. BMC Bioinform. 2022, 23, 135. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Wu, W.; Xie, C.; Zhao, G.; Zhao, Y.; Chen, R. NPInter v2.0: An updated database of ncRNA interactions. Nucleic Acids Res. 2014, 42, D104–D108. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [Green Version]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [Green Version]
- Neph, S.; Kuehn, M.S.; Reynolds, A.P.; Haugen, E.; Thurman, R.E.; Johnson, A.K.; Rynes, E.; Maurano, M.T.; Vierstra, J.; Thomas, S.; et al. BEDOPS: High-performance genomic feature operations. Bioinformatics 2012, 28, 1919–1920. [Google Scholar] [CrossRef] [Green Version]
- Flicek, P.; Amode, M.R.; Barrell, D.; Beal, K.; Brent, S.; Chen, Y.; Clapham, P.; Coates, G.; Fairley, S.; Fitzgerald, S.; et al. Ensembl 2011. Nucleic Acids Res. 2011, 39, D800–D806. [Google Scholar] [CrossRef]
- Frank, E.; Hall, M.A.; Witten, I.H. The WEKA Workbench. In Data Mining: Practical Machine Learning Tools and Techniques, 4th ed.; Morgan Kaufmann: Burlington, MA, USA, 2016. [Google Scholar]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: AnRPackage for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Cui, Y.; Yao, Y.; An, L.; Bai, Y.; Li, X.; Yao, X.; Wu, K. Genome-wide identification of WD40 transcription factors and their regulation of the MYB-bHLH-WD40 (MBW) complex related to anthocyanin synthesis in Qingke (Hordeum vulgare L. var. nudum Hook. f.). BMC Genom. 2023, 24, 166. [Google Scholar] [CrossRef] [PubMed]
- Buhrman, K.; Aravena-Calvo, J.; Zaulich, C.R.; Hinz, K.; Laursen, T. Anthocyanic Vacuolar Inclusions: From Biosynthesis to Storage and Possible Applications. Front. Chem. 2022, 10, 913324. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Ru, Y.; Chuang, L.-S.; Hsu, N.-Y.; Shi, L.-S.; Hakenberg, J.; Cheng, W.-Y.; Uzilov, A.; Ding, W.; Glicksberg, B.S.; et al. Disease-associated variants in different categories of disease located in distinct regulatory elements. BMC Genom. 2015, 16 (Suppl. 8), S3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrile, M.C.; Tebez, N.M.; Colman, S.L.; Mateos, J.L.; Morato-López, E.; Sánchez-López, N.; Izquierdo-Álvarez, A.; Marina, A.; Villalobos, L.I.A.C.; Estelle, M.; et al. S-Nitrosation of E3 Ubiquitin Ligase Complex Components Regulates Hormonal Signalings in Arabidopsis. Front. Plant Sci. 2022, 12, 794582. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Gu, J.; Yao, J.; Li, Y.; Zhang, Z.; Xia, W.; Wang, Z.; Gui, X.; Li, L.; Li, D.; et al. Liquid-liquid phase separation of RBGD2/4 is required for heat stress resistance in Arabidopsis. Dev. Cell 2022, 57, 583–597.e6. [Google Scholar] [CrossRef]
- Feng, C.; Cai, X.-W.; Su, Y.-N.; Li, L.; Chen, S.; He, X.-J. Arabidopsis RPD3-like histone deacetylases form multiple complexes involved in stress response. J. Genet. Genom. 2021, 48, 369–383. [Google Scholar] [CrossRef]
- Truebestein, L.; Leonard, T.A. Coiled-coils: The long and short of it. Bioessays 2016, 38, 903–916. [Google Scholar] [CrossRef] [Green Version]
- Dang, M.; Li, T.; Song, J. ATP and nucleic acids competitively modulate LLPS of the SARS-CoV2 nucleocapsid protein. Commun. Biol. 2023, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.; Li, T.; Zhou, S.; Song, J. Arg/Lys-containing IDRs are cryptic binding domains for ATP and nucleic acids that interplay to modulate LLPS. Commun. Biol. 2022, 5, 1315. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, M.; Duncan, S.; Yang, X.; Abdelhamid, M.; Huang, L.; Zhang, H.; Benfey, P.N.; Waller, Z.A.E.; Ding, Y. G-quadruplex structures trigger RNA phase separation. Nucleic Acids Res. 2019, 47, 11746–11754. [Google Scholar] [CrossRef]
- Langdon, E.M.; Gladfelter, A.S. Chapter Four—Probing RNA Structure in Liquid–Liquid Phase Separation Using SHAPE-MaP. In Methods in Enzymology; Rhoades, E., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 611, pp. 67–79. [Google Scholar] [CrossRef]
- Zhu, H.; Fu, H.; Cui, T.; Ning, L.; Shao, H.; Guo, Y.; Ke, Y.; Zheng, J.; Lin, H.; Wu, X.; et al. RNAPhaSep: A resource of RNAs undergoing phase separation. Nucleic Acids Res. 2022, 50, D340–D346. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Amino Acid Group Letter | Amino Acid | Amino Acid Features |
|---|---|---|
| P | R, K, S, T | Positive or polar uncharged |
| N | D, E, N, Q | Negative or polar uncharged |
| H | A, V, I, L, M | Hydrophobic |
| R | F, W, Y | Ring structures |
| S | C, G, P, H | Special properties |
| DNA Group Letter | DNA/Ambiguous DNA |
|---|---|
| G | G |
| Z | R, S, K, B, D, V |
| X | A, C, T, Y, W, M, H |
| N | N |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, J.E.; Jun, J.H.; Kwon, J.H.; Lee, J.-H.; Hwang, K.; Kim, S.; Jeong, N. Arabidopsis Transcription Regulatory Factor Domain/Domain Interaction Analysis Tool—Liquid/Liquid Phase Separation, Oligomerization, GO Analysis: A Toolkit for Interaction Data-Based Domain Analysis. Genes 2023, 14, 1476. https://doi.org/10.3390/genes14071476
Kang JE, Jun JH, Kwon JH, Lee J-H, Hwang K, Kim S, Jeong N. Arabidopsis Transcription Regulatory Factor Domain/Domain Interaction Analysis Tool—Liquid/Liquid Phase Separation, Oligomerization, GO Analysis: A Toolkit for Interaction Data-Based Domain Analysis. Genes. 2023; 14(7):1476. https://doi.org/10.3390/genes14071476
Chicago/Turabian StyleKang, Jee Eun, Ji Hae Jun, Jung Hyun Kwon, Ju-Hyun Lee, Kidong Hwang, Sungjong Kim, and Namhee Jeong. 2023. "Arabidopsis Transcription Regulatory Factor Domain/Domain Interaction Analysis Tool—Liquid/Liquid Phase Separation, Oligomerization, GO Analysis: A Toolkit for Interaction Data-Based Domain Analysis" Genes 14, no. 7: 1476. https://doi.org/10.3390/genes14071476
APA StyleKang, J. E., Jun, J. H., Kwon, J. H., Lee, J.-H., Hwang, K., Kim, S., & Jeong, N. (2023). Arabidopsis Transcription Regulatory Factor Domain/Domain Interaction Analysis Tool—Liquid/Liquid Phase Separation, Oligomerization, GO Analysis: A Toolkit for Interaction Data-Based Domain Analysis. Genes, 14(7), 1476. https://doi.org/10.3390/genes14071476