
Evidence for Two Soybean Looper Strains in the United States with Limited Capacity for Cross-Hybridization

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimen Collections
2.2. DNA Preparation
2.3. Isolation of the DNA Segments by PCR
2.4. Characterization of Haplotypes
2.5. DNA Sequence Analysis, Statistics, and Data Availability
3. Results
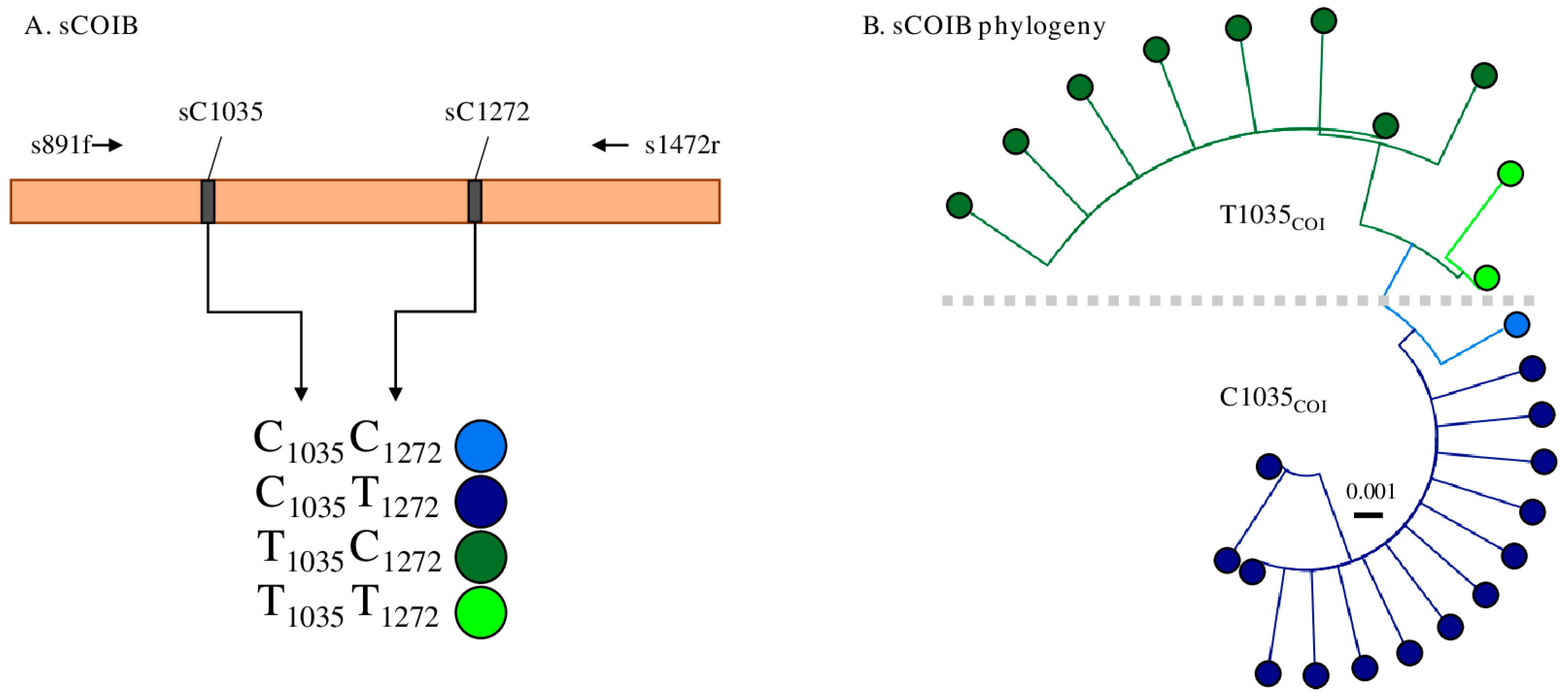
3.1. Characterization of the Mitochondrial sCOIB Segment
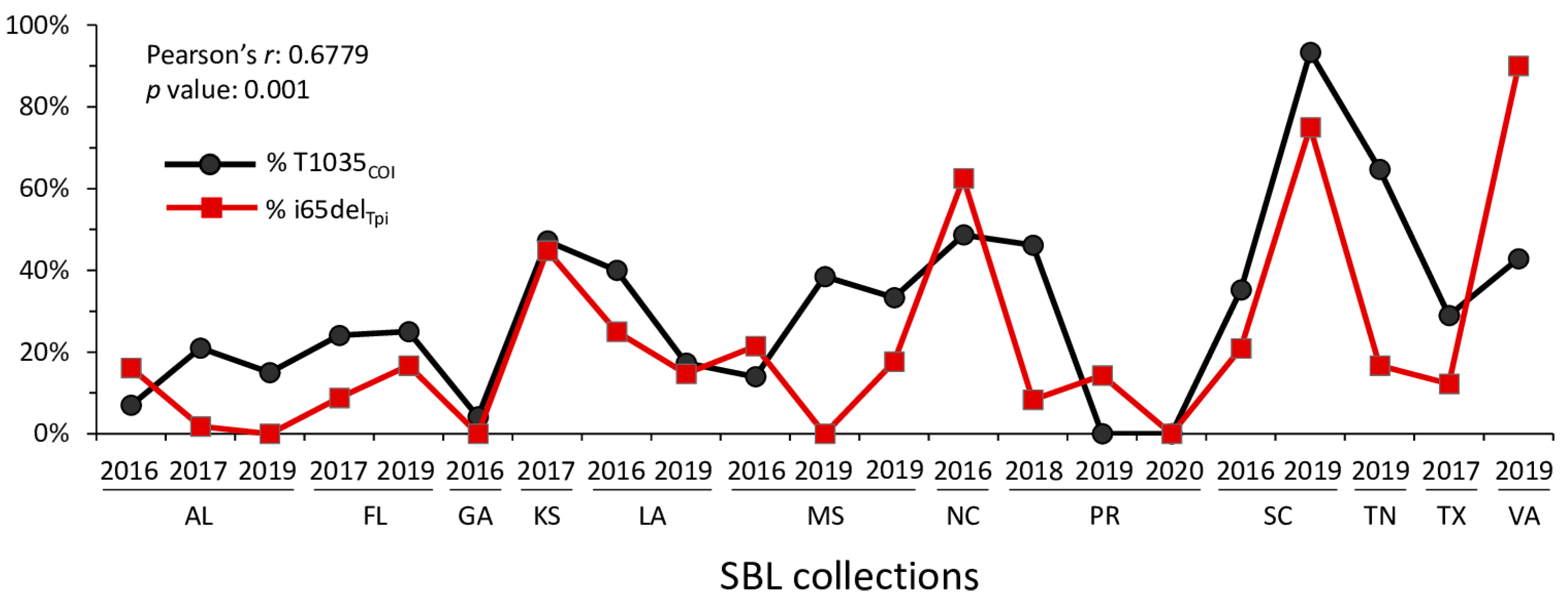
3.2. Linkage between i65delTpi and T1035COI
3.3. Frequency of i65del/+ Heterozygotes
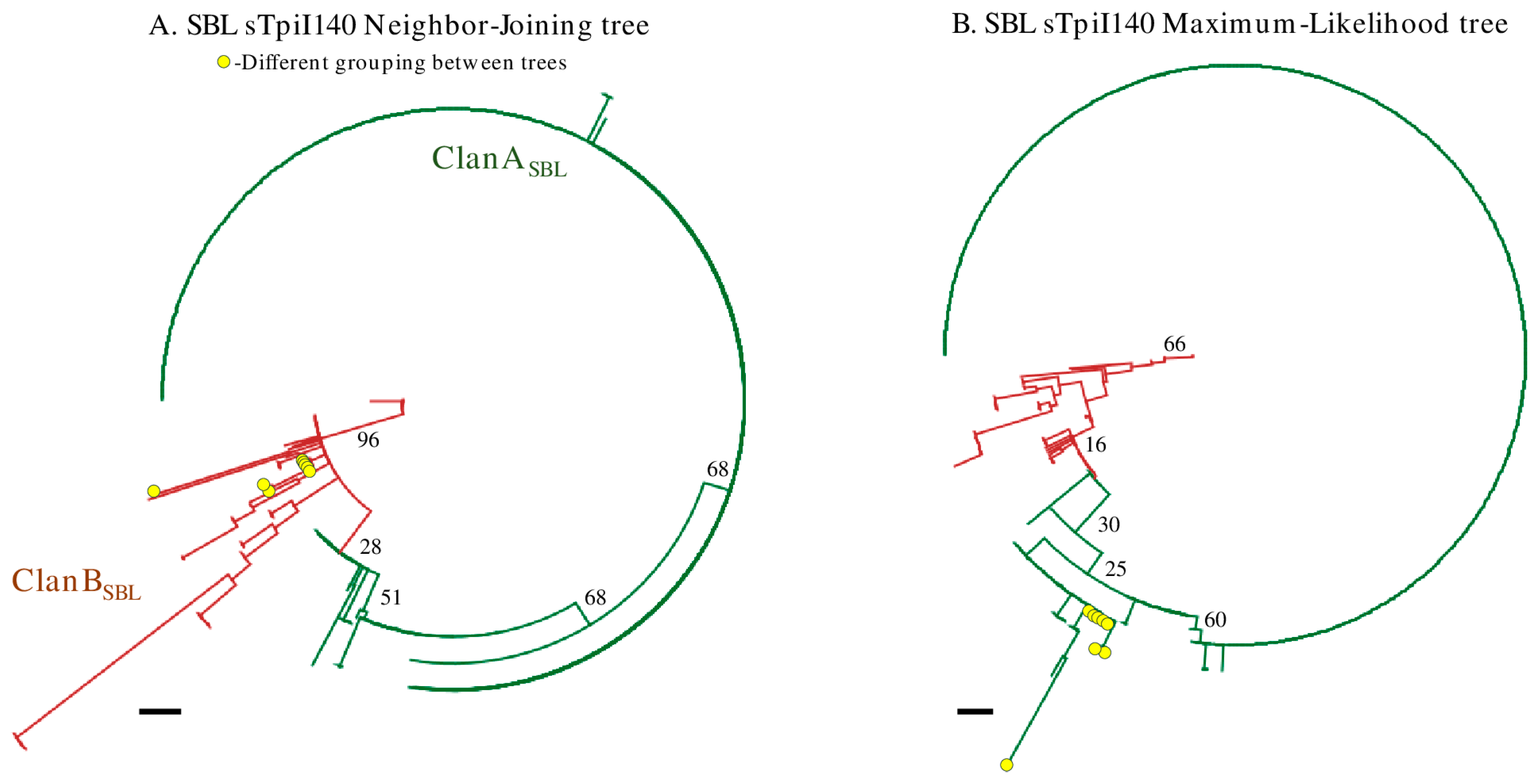
3.4. The Phylogeny of the sTpiI140 Segment
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vandermeer, J.; Pascual, M. Competitive coexistence through intermediate polyphagy. Ecol. Complex. 2006, 3, 37–43. [Google Scholar] [CrossRef]
- Stireman, J.O. The evolution of generalization? Parasitoid flies and the perils of inferring host range evolution from phylogenies. J. Evol. Biol. 2005, 18, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Specht, A.; de Paula-Moraes, S.V.; Sosa-Gomez, D.R. Host plants of Chrysodeixis includens (Walker) (Lepidoptera, Noctuidae, Plusiinae). Rev. Bras. Entomol. 2015, 59, 343–345. [Google Scholar] [CrossRef]
- Luginbill, P. The Fall Armyworm; U.S. Department of Agriculture Technical Bulletins: Philadelphia, PA, USA, 1928; Volume 34, pp. 1–91. [Google Scholar]
- Montezano, D.G.; Specht, A.; Sosa-Gomez, D.R.; Roque-Specht, V.F.; Sousa-Silva, J.C.; Paula-Moraes, S.V.; Peterson, J.A.; Hunt, T.E. Host plants of Spodoptera frugiperda (Lepidoptera: Noctuidae) in the Americas. Afr. Entomol. 2018, 26, 286–300. [Google Scholar] [CrossRef] [Green Version]
- Groot, A.T.; Unbehend, M.; Hänniger, S.; Juárez, M.L.; Kost, S.; Heckel, D.G. Evolution of reproductive isolation of Spodoptera frugiperda. In Pheromone Communication in Moths: Evolution, Behavior, and Application; Jeremy, D.A., Ring, T.C., Eds.; University of California Press: Berkeley, CA, USA, 2016. [Google Scholar]
- Nagoshi, R.N.; Meagher, R.L. The Spodoptera frugiperda host strains: What they are and why they matter for understanding and controlling this global agricultural pest. J. Econ. Entomol. 2022, 115, 1729–1743. [Google Scholar] [CrossRef]
- Martin, S.H.; Most, M.; Palmer, W.J.; Salazar, C.; McMillan, W.O.; Jiggins, F.M.; Jiggins, C.D. Natural selection and genetic diversity in the butterfly Heliconius melpomene. Genetics 2016, 203, 525–541. [Google Scholar] [CrossRef] [Green Version]
- Palma, J.; Maebe, K.; Guedes, J.V.C.; Smagghe, G. Molecular variability and genetic structure of Chrysodeixis includens (Lepidoptera: Noctuidae), an important soybean defoliator in Brazil. PLoS ONE 2015, 10, e0121260. [Google Scholar] [CrossRef] [Green Version]
- Silva, C.S.; Cordeiro, E.M.G.; de Paiva, J.B.; Dourado, P.M.; Carvalho, R.A.; Head, G.; Martinelli, S.; Correa, A.S. Population expansion and genomic adaptation to agricultural environments of the soybean looper, Chrysodeixis includens. Evol. Appl. 2020, 13, 2071–2085. [Google Scholar] [CrossRef] [Green Version]
- Coyne, J.A.; Orr, H.A. Patterns of Speciation in Drosophila. Evolution 1989, 43, 362–381. [Google Scholar] [CrossRef]
- Haldane, J.B.S. Sex ratio and unisexual sterility in hybrid animals. J. Genet. 1922, 12, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Oppenheim, S.; Hopper, K. Genetics of host range in Lepidoptera. In Molecular Biology and Genetics of the Lepidoptera; Goldsmith, M.R., Marec, F., Eds.; CRC Press: Boca Raton, FL, USA, 2010; pp. 195–217. [Google Scholar]
- Sperling, F.A.H. Sex-linked genes and species-differences in Lepidoptera. Can. Entomol. 1994, 126, 807–818. [Google Scholar] [CrossRef]
- Prowell, D.P. Sex linkage and speciation in Lepidoptera. In Endless Forms: Species and Speciation; Howard, D., Berlocher, S., Eds.; Oxford Press: New York, NY, USA, 1998; pp. 309–319. [Google Scholar]
- Nagoshi, R.N. The fall armyworm Triosephosphate isomerase (Tpi) gene as a marker of strain identity and interstrain mating. Ann. Entomol. Soc. Am. 2010, 103, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Nagoshi, R.N.; Meagher, R.L. Using intron sequence comparisons in the Triose-phosphate isomerase gene to study the divergence of the fall armyworm host strains. Insect Mol. Biol. 2016, 25, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Dopman, E.B.; Bogdanowicz, S.M.; Harrison, R.G. Genetic mapping of sexual isolation between E and Z pheromone strains of the European corn borer (Ostrinia nubilalis). Genetics 2004, 167, 301–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagoshi, R.N.; Meagher, R.L.; Flanders, K.; Gore, J.; Jackson, R.; Lopez, J.; Armstrong, J.S.; Buntin, G.D.; Sansone, C.; Leonard, B.R. Using haplotypes to monitor the migration of fall armyworm (Lepidoptera: Noctuidae) corn-strain populations from Texas and Florida. J. Econ. Entomol. 2008, 101, 742–749. [Google Scholar] [CrossRef]
- Nagoshi, R.N. Observations of genetic differentiation between the fall armyworm host strains. PLoS ONE 2022, 17, e0277510. [Google Scholar] [CrossRef]
- Nagoshi, R.N.; Vizuete, J.L.A.; Murua, M.G.; Garces-Carrera, S. Comparisons of fall armyworm haplotypes between the Galapagos Islands and mainland Ecuador indicate limited migration to and between islands. Sci. Rep. 2021, 11, 3457. [Google Scholar] [CrossRef]
- Drummond, A.J.; Ashton, B.; Buxton, S.; Cheung, M.; Cooper, A.; Duran, C.; Field, M.; Heled, J.; Kearse, M.; Markowitz, S.; et al. Geneious v5.5; Geneious: Auckland, New Zealand, 2010. [Google Scholar]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef]
- Nagoshi, R.N.; Davis, J.A.; Meagher, R.L.; Musser, F.R.; Head, G.P.; Portillo, H.; Teran, H. Evidence for two soybean looper strains in the United States with limited capacity for cross-hybridization. Genes 2023. submitted. [Google Scholar]
- Garnier-Géré, P.; Chikhi, L. Population Subdivision, Hardy–Weinberg Equilibrium and the Wahlund Effect. In The Encyclopedia of Life; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Collection | State | Year | Type | Collector |
|---|---|---|---|---|
| A. Field collections | ||||
| AL2017 | Alabama | 2017 | Pheromone trap | R. Meagher |
| FL2017 | Florida | 2017 | Pheromone trap | R. Meagher |
| FL2019 | Florida | 2019 | Pheromone trap | R. Meagher |
| MS2019a | Mississippi | 2019 | Pheromone trap | R. Meagher |
| TX2017 | Texas | 2017 | Pheromone trap | R. Parker |
| KS2017 | Kansas | 2017 | Pheromone trap | B. McCornack |
| PR2018 | Puerto Rico | 2018 | Larvae, field | H. Portillo |
| PR2019 | Puerto Rico | 2019 | Larvae, field | H. Teran |
| B. Colony collections | ||||
| AL2016 | Alabama | 2016 | F1 generation | J. Davis |
| GA2016 | Georgia | 2016 | F1 generation | J. Davis |
| LA2016 | Louisiana | 2016 | F1 generation | J. Davis |
| MS2016 | Mississippi | 2016 | F1 generation | J. Davis |
| NC2016 | N. Carolina | 2016 | F1 generation | J. Davis |
| SC2016 | S. Carolina | 2016 | F1 generation | J. Davis |
| AL2019 | Alabama | 2019 | F1-2 generation | F. Musser, B. Catchot |
| LA2019 | Louisiana | 2019 | F1-2 generation | F. Musser, B. Catchot |
| MS2019b | Mississippi | 2019 | F1-2 generation | F. Musser, B. Catchot |
| PR2020 | Puerto Rico | 2020 | F1-2 generation | F. Musser, B. Catchot |
| SC2019 | S. Carolina | 2019 | F1-2 generation | F. Musser, B. Catchot |
| TN2019 | Tennessee | 2019 | F1-2 generation | F. Musser, B. Catchot |
| VA2019 | Virginia | 2019 | F1-2 generation | F. Musser, B. Catchot |
| i65delTpi Frequency (%) | t-Test Scores | |||
|---|---|---|---|---|
| Collection | n | C1035COI | T1035COI | p (t, df) |
| A. Mean Colony 1 | 410 | 18 ± 5 | 47 ± 11 | p = 0.007 * (5.203, 4) |
| B. Mean Field 2 | 533 | 18 ± 5 | 27 ± 11 | p = 0.037 * (3.075, 4) |
| C. Colony + Field | ||||
| AL | 144 | 16 | 26 | |
| FL | 190 | 14 | 46 | |
| KS | 53 | 27 | 42 | |
| LA | 92 | 13 | 54 | |
| MS | 80 | 24 | 29 | |
| NC | 37 | 21 | 44 | |
| SC | 86 | 14 | 36 | |
| TX | 145 | 20 | 42 | |
| GA-VA-TN-PR | 116 | 13 | 48 | |
| Mean ± sd | 18 ± 5 | 41 ± 9 | p = 0.004 * (5.808, 8) | |
| A. Male adults | Genotype (number) | |||
| Collection | i65del/i65del | +/+ | i65del/+ | Fis |
| AL2017 | 1 | 56 | 25 | −0.11 |
| FL2017 | 11 | 125 | 26 | 0.36 |
| FL2019 | 4 | 24 | 0 | 1.00 |
| KS2017 | 13 | 29 | 11 | 0.54 |
| MS2019a | 3 | 17 | 4 | 0.49 |
| TX2017 | 11 | 90 | 44 | 0.14 |
| Total | 43 | 341 | 109 | 0.30 |
| Mean | 0.41 | |||
| B. Larvae of unknown gender | ||||
| Collection | i65del/i65del or i65del/W | +/+ or +/W | i65del/+ | Fis |
| AL2016 | 5 | 31 | 7 | 0.49 |
| GA2016 | 0 | 24 | 0 | nd 1 |
| LA2016 | 7 | 28 | 5 | 0.65 |
| LA2019 | 5 | 34 | 13 | 0.27 |
| MS2016 | 6 | 28 | 9 | 0.43 |
| NC2016 | 10 | 16 | 11 | 0.39 |
| PR2019 | 3 | 21 | 3 | 0.60 |
| SC2016 | 9 | 43 | 19 | 0.31 |
| VA2019 | 9 | 10 | 2 | 0.81 |
| Total | 54 | 235 | 69 | 0.48 |
| Mean | 0.49 | |||
| Metric | Tree Type | ClanA | ClanB |
|---|---|---|---|
| Tpi marker: i65delTpi | NJ | 0% | 100% |
| ML | 2% | 98% | |
| COI marker: T1035COI | NJ | 17% | 41% |
| ML | 18% | 41% | |
| Haplotype diversity (±sd) | NJ | 0.229 ± 0.025 | 0.935 ± 0.014 |
| ML | 0.194 ± 0.024 | 0.943 ± 0.011 | |
| Nucleotide diversity (±sd) | NJ | 0.003 ± 0.000 | 0.024 ± 0.144 |
| ML | 0.002 ± 0.001 | 0.026 ± 0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagoshi, R.N.; Davis, J.A.; Meagher, R.L.; Musser, F.R.; Head, G.P.; Portillo, H.; Teran, H. Evidence for Two Soybean Looper Strains in the United States with Limited Capacity for Cross-Hybridization. Genes 2023, 14, 1509. https://doi.org/10.3390/genes14071509
Nagoshi RN, Davis JA, Meagher RL, Musser FR, Head GP, Portillo H, Teran H. Evidence for Two Soybean Looper Strains in the United States with Limited Capacity for Cross-Hybridization. Genes. 2023; 14(7):1509. https://doi.org/10.3390/genes14071509
Chicago/Turabian StyleNagoshi, Rodney N., Jeffrey A. Davis, Robert L. Meagher, Fred R. Musser, Graham P. Head, Hector Portillo, and Henry Teran. 2023. "Evidence for Two Soybean Looper Strains in the United States with Limited Capacity for Cross-Hybridization" Genes 14, no. 7: 1509. https://doi.org/10.3390/genes14071509
APA StyleNagoshi, R. N., Davis, J. A., Meagher, R. L., Musser, F. R., Head, G. P., Portillo, H., & Teran, H. (2023). Evidence for Two Soybean Looper Strains in the United States with Limited Capacity for Cross-Hybridization. Genes, 14(7), 1509. https://doi.org/10.3390/genes14071509