
Sodium Channel Gene Variants in Fetuses with Abnormal Sonographic Findings: Expanding the Prenatal Phenotypic Spectrum of Sodium Channelopathies
 , , , ,
, , , , {kind=link}
{kind=link}
Abstract
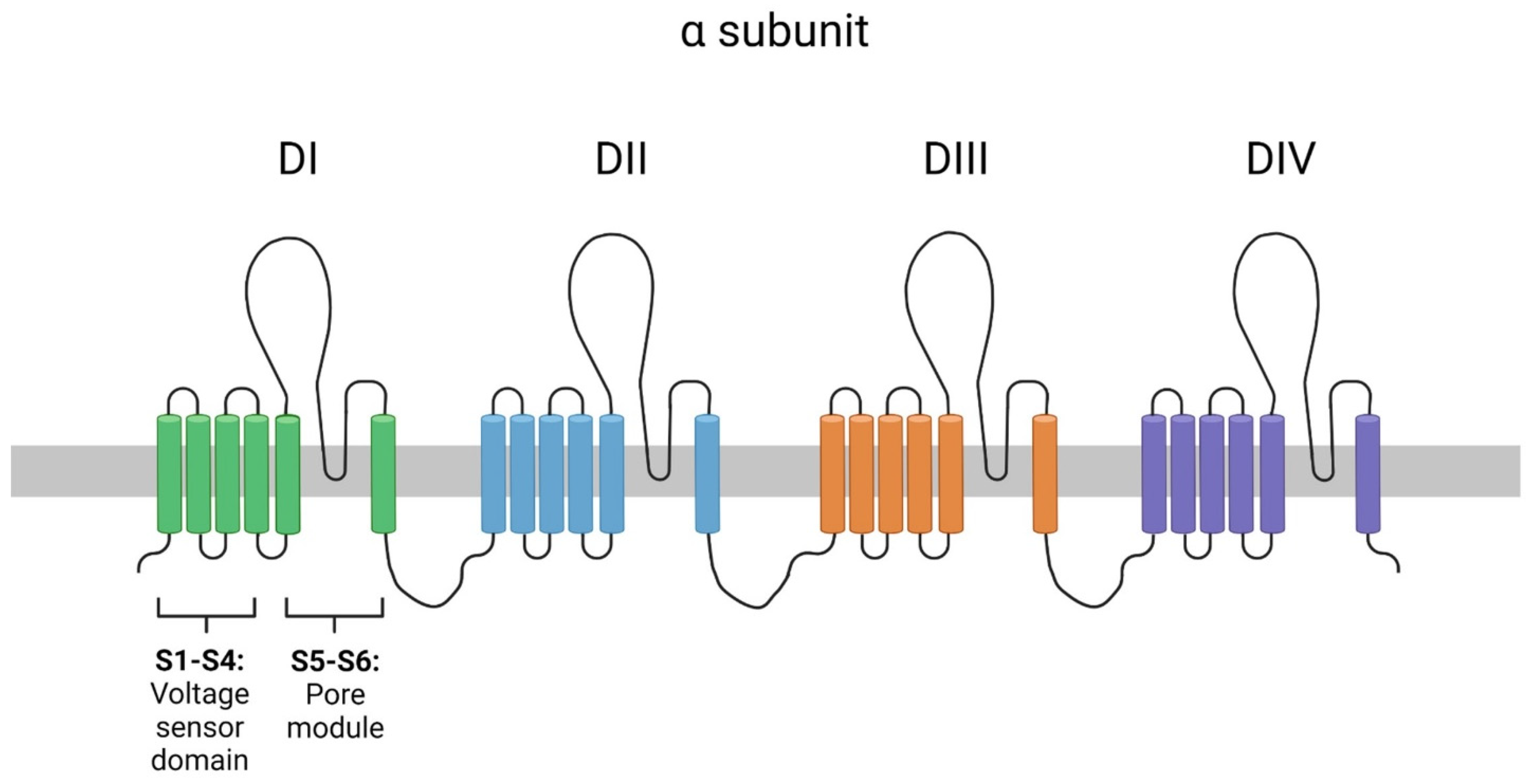
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.1.1. Family 1
2.1.2. Family 2
2.2. Genetic Investigation
3. Results
3.1. Family 1
3.2. Family 2
4. Discussion
4.1. Family 1
4.2. Family 2
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ogiwara, I.; Ito, K.; Sawaishi, Y.; Osaka, H.; Mazaki, E.; Inoue, I.; Montal, M.; Hashikawa, T.; Shike, T.; Fujiwara, T.; et al. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology 2009, 73, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Meisler, M.H.; Hill, S.F.; Yu, W. Sodium channelopathies in neurodevelopmental disorders. Nat. Rev. Neurosci. 2021, 22, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Matalon, D.; Goldberg, E.; Medne, L.; Marsh, E.D. Confirming an expanded spectrum of SCN2A mutations: A case series. Epileptic Disord. 2014, 16, 13–18. [Google Scholar] [CrossRef]
- Munroe, P.B.; Addison, S.; Abrams, D.J.; Sebire, N.J.; Cartwright, J.; Donaldson, I.; Cohen, M.M.; Mein, C.; Tinker, A.; Harmer, S.C.; et al. Postmortem Genetic Testing for Cardiac Ion Channelopathies in Stillbirths. Circ. Genom. Precis. Med. 2018, 11, e001817. [Google Scholar] [CrossRef]
- Tsujino, A.; Maertenst, C.; Ohno, K.; Shen, X.M.; Fukuda, T.; Harper, C.M.; Cannon, S.C.; Engel, A.G. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc. Natl. Acad. Sci. USA 2003, 100, 7377–7382. [Google Scholar] [CrossRef]
- Kilby, M.D. The role of next-generation sequencing in the investigation of ultrasound-identified fetal structural anomalies. BJOG Int. J. Obstet. Gynaecol. 2021, 128, 420–429. [Google Scholar] [CrossRef]
- Jelin, A.C.; Vora, N. Whole Exome Sequencing: Applications in Prenatal Genetics. Obstet. Gynecol. Clin. N. Am. 2018, 15, 69–81. [Google Scholar] [CrossRef]
- Fu, F.; Li, R.; Yu, Q.; Wang, D.; Deng, Q.; Li, L.; Lei, T.; Chen, G.; Nie, Z.; Yang, X.; et al. Application of exome sequencing for prenatal diagnosis of fetal structural anomalies: Clinical experience and lessons learned from a cohort of 1618 fetuses. Genome Med. 2022, 14, 123. [Google Scholar] [CrossRef] [PubMed]
- Rajcan-Separovic, E. Next generation sequencing in recurrent pregnancy loss-approaches and outcomes. Eur. J. Med. Genet. 2020, 63, 103644. [Google Scholar] [CrossRef]
- Janicki, E.; De Rademaeker, M.; Meunier, C.; Boeckx, N.; Blaumeiser, B.; Janssens, K. Implementation of Exome Sequencing in Prenatal Diagnostics: Chances and Challenges. Diagnostics 2023, 13, 860. [Google Scholar] [CrossRef]
- Mone, F.; Abu Subieh, H.; Doyle, S.; Hamilton, S.; Mcmullan, D.J.; Allen, S.; Marton, T.; Williams, D.; Kilby, M.D. Evolving fetal phenotypes and clinical impact of progressive prenatal exome sequencing pathways: Cohort study. Ultrasound Obstet. Gynecol. 2022, 59, 723–730. [Google Scholar] [CrossRef]
- Polipalli, S.K.; Karra, V.K.; Jindal, A.; Puppala, M.; Singh, P.; Rawat, K.; Kapoor, S. Cytogenetic analysis for suspected chromosomal abnormalities; A five years experience. J. Clin. Diagn. Res. 2016, 10, GC01–GC05. [Google Scholar] [CrossRef] [PubMed]
- Schuler, B.A.; Nelson, E.T.; Koziura, M.; Cogan, J.D.; Hamid, R.; Phillips, J.A. Lessons learned: Next-generation sequencing applied to undiagnosed genetic diseases. J. Clin. Investig. 2022, 132, e154942. [Google Scholar] [CrossRef] [PubMed]
- Dhombres, F.; Morgan, P.; Chaudhari, B.P.; Filges, I.; Sparks, T.N.; Lapunzina, P.; Roscioli, T.; Agarwal, U.; Aggarwal, S.; Beneteau, C.; et al. Prenatal phenotyping: A community effort to enhance the Human Phenotype Ontology. Am. J. Med. Genet. Part C Semin. Med. Genet. 2022, 190, 231–242. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child. Educ. Pract. Ed. 2013, 98, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Normand, E.A.; Braxton, A.; Nassef, S.; Ward, P.A.; Vetrini, F.; He, W.; Patel, V.; Qu, C.; Westerfield, L.E.; Stover, S.; et al. Clinical exome sequencing for fetuses with ultrasound abnormalities and a suspected Mendelian disorder. Genome Med. 2018, 10, 74. [Google Scholar] [CrossRef]
- Monaghan, K.G.; Leach, N.T.; Pekarek, D.; Prasad, P.; Rose, N.C. The use of fetal exome sequencing in prenatal diagnosis: A points to consider document of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2020, 22, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Mellis, R.; Oprych, K.; Scotchman, E.; Hill, M.; Chitty, L.S. Diagnostic yield of exome sequencing for prenatal diagnosis of fetal structural anomalies: A systematic review and meta-analysis. Prenat. Diagn. 2022, 42, 662–685. [Google Scholar] [CrossRef]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The human phenotype ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1317. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Ellard, S.; Baple, E.L.; Callaway, A.; Berry, I.; Forrester, N.; Turnbull, C.; Owens, M.; Eccles, D.M.; Abbs, S.; Scott, R.; et al. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020. 2020. Available online: https://www.acgs.uk.com/media/12443/uk-practice-guidelines-for-variant-classification-v1-2023.pdf (accessed on 20 December 2023).
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. OMIM.org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019, 47, D1038–D1043. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Quereda, L.; Rodriguez, M.J.; Diaz-Manera, J.; Alonso-Perez, J.; Gallardo, E.; Nascimento, A.; Ortez, C.; Natera-De Benito, D.; Olive, M.; Gonzalez-Mera, L.; et al. Targeted next-generation sequencing in a large cohort of genetically undiagnosed patients with neuromuscular disorders in Spain. Genes 2020, 11, 539. [Google Scholar] [CrossRef] [PubMed]
- Zaharieva, I.T.; Thor, M.G.; Oates, E.C.; Van Karnebeek, C.; Hendson, G.; Blom, E.; Witting, N.; Rasmussen, M.; Gabbett, M.T.; Ravenscroft, G.; et al. Loss-of-function mutations in SCN4A cause severe foetal hypokinesia or “classical” congenital myopathy. Brain 2016, 139, 674–691. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Yang, Y.; Duan, J.; Niu, X.; Chen, Y.; Wang, D.; Zhang, J.; Chen, J.; Yang, X.; Li, J.; et al. SCN2A-Related Epilepsy: The Phenotypic Spectrum, Treatment and Prognosis. Front. Mol. Neurosci. 2022, 15, 809951. [Google Scholar] [CrossRef]
- Li, M.; Jancovski, N.; Jafar-Nejad, P.; Burbano, L.E.; Rollo, B.; Richards, K.; Drew, L.; Sedo, A.; Heighway, J.; Pachernegg, S.; et al. Antisense oligonucleotide therapy reduces seizures and extends life span in an SCN2A gain-of-function epilepsy model. J. Clin. Investig. 2021, 131, e152079. [Google Scholar] [CrossRef]
- Scalmani, P.; Rusconi, R.; Armatura, E.; Zara, F.; Avanzini, G.; Franceschetti, S.; Mantegazza, M. Effects in neocortical neurons of mutations of the Nav1.2 Na+ channel causing benign familial neonatal-infantile seizures. J. Neurosci. 2006, 26, 10100–10109. [Google Scholar] [CrossRef]
- Wolff, M.; Johannesen, K.M.; Hedrich, U.B.S.; Masnada, S.; Rubboli, G.; Gardella, E.; Lesca, G.; Ville, D.; Milh, M.; Villard, L.; et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017, 140, 1316–1336. [Google Scholar] [CrossRef]
- Lauxmann, S.; Boutry-Kryza, N.; Rivier, C.; Mueller, S.; Hedrich, U.B.S.; Maljevic, S.; Szepetowski, P.; Lerche, H.; Lesca, G. An SCN2A mutation in a family with infantile seizures from Madagascar reveals an increased subthreshold Na+ current. Epilepsia 2013, 54, e117–e121. [Google Scholar] [CrossRef]
- Liao, Y.; Anttonen, A.K.; Liukkonen, E.; Gaily, E.; Maljevic, S.; Schubert, S.; Bellan-Koch, A.; Petrou, S.; Ahonen, V.E.; Lerche, H.; et al. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology 2010, 75, 1454–1458. [Google Scholar] [CrossRef]
- Kruth, K.A.; Grisolano, T.M.; Ahern, C.A.; Williams, A.J. SCN2A channelopathies in the autism spectrum of neuropsychiatric disorders: A role for pluripotent stem cells? Mol. Autism 2020, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Howell, K.B.; McMahon, J.M.; Carvill, G.L.; Tambunan, D.; Mackay, M.T.; Rodriguez-Casero, V.; Webster, R.; Clark, D.; Freeman, J.L.; Calvert, S.; et al. SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures. Neurology 2015, 85, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Dhamija, R.; Wirrell, E.; Falcao, G.; Kirmani, S.; Wong-Kisiel, L.C. Novel de novo SCN2A mutation in a child with migrating focal seizures of infancy. Pediatr. Neurol. 2013, 49, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, S.; Marchionni, E.; Prudente, S.; De Liso, P.; Spalice, A.; Giancotti, A.; Manganaro, L.; Pizzuti, A. Unusual association of SCN2A epileptic encephalopathy with severe cortical dysplasia detected by prenatal MRI. Eur. J. Paediatr. Neurol. 2017, 21, 587–590. [Google Scholar] [CrossRef]
- Tzialla, C.; Arossa, A.; Mannarino, S.; Orcesi, S.; Veggiotti, P.; Fiandrino, G.; Zuffardi, O.; Errichiello, E. SCN2A and arrhythmia: A potential correlation? A case report and literature review. Eur. J. Med. Genet. 2022, 65, 104639. [Google Scholar] [CrossRef]
- Cannon, S.C. Sodium channelopathies of skeletal muscle. In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2018; Volume 246, pp. 309–330. [Google Scholar] [CrossRef]
- Elia, N.; Palmio, J.; Castañeda, M.S.; Shieh, P.B.; Quinonez, M.; Suominen, T.; Hanna, M.G.; Männikkö, R.; Udd, B.; Cannon, S.C. Myasthenic congenital myopathy from recessive mutations at a single residue in NaV1.4. Neurology 2019, 92, e1405–e1415. [Google Scholar] [CrossRef]
- Flood, E.; Boiteux, C.; Allen, T.W. Selective ion permeation involves complexation with carboxylates and lysine in a model human sodium channel. PLoS Comput. Biol. 2018, 14, e1006398. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hadjipanteli, A.; Theodosiou, A.; Papaevripidou, I.; Evangelidou, P.; Alexandrou, A.; Salameh, N.; Kallikas, I.; Kakoullis, K.; Frakala, S.; Oxinou, C.; et al. Sodium Channel Gene Variants in Fetuses with Abnormal Sonographic Findings: Expanding the Prenatal Phenotypic Spectrum of Sodium Channelopathies. Genes 2024, 15, 119. https://doi.org/10.3390/genes15010119
Hadjipanteli A, Theodosiou A, Papaevripidou I, Evangelidou P, Alexandrou A, Salameh N, Kallikas I, Kakoullis K, Frakala S, Oxinou C, et al. Sodium Channel Gene Variants in Fetuses with Abnormal Sonographic Findings: Expanding the Prenatal Phenotypic Spectrum of Sodium Channelopathies. Genes. 2024; 15(1):119. https://doi.org/10.3390/genes15010119
Chicago/Turabian StyleHadjipanteli, Andrea, Athina Theodosiou, Ioannis Papaevripidou, Paola Evangelidou, Angelos Alexandrou, Nicole Salameh, Ioannis Kallikas, Kyriakos Kakoullis, Sofia Frakala, Christina Oxinou, and et al. 2024. "Sodium Channel Gene Variants in Fetuses with Abnormal Sonographic Findings: Expanding the Prenatal Phenotypic Spectrum of Sodium Channelopathies" Genes 15, no. 1: 119. https://doi.org/10.3390/genes15010119
APA StyleHadjipanteli, A., Theodosiou, A., Papaevripidou, I., Evangelidou, P., Alexandrou, A., Salameh, N., Kallikas, I., Kakoullis, K., Frakala, S., Oxinou, C., Marnerides, A., Kousoulidou, L., Anastasiadou, V. C., & Sismani, C. (2024). Sodium Channel Gene Variants in Fetuses with Abnormal Sonographic Findings: Expanding the Prenatal Phenotypic Spectrum of Sodium Channelopathies. Genes, 15(1), 119. https://doi.org/10.3390/genes15010119