
A Study of Polish Family with Scoliosis and Limb Contractures Expands the MYH3 Disease Spectrum

 , , , , and
, , , , and 
Abstract
1. Introduction
2. Materials and Methods
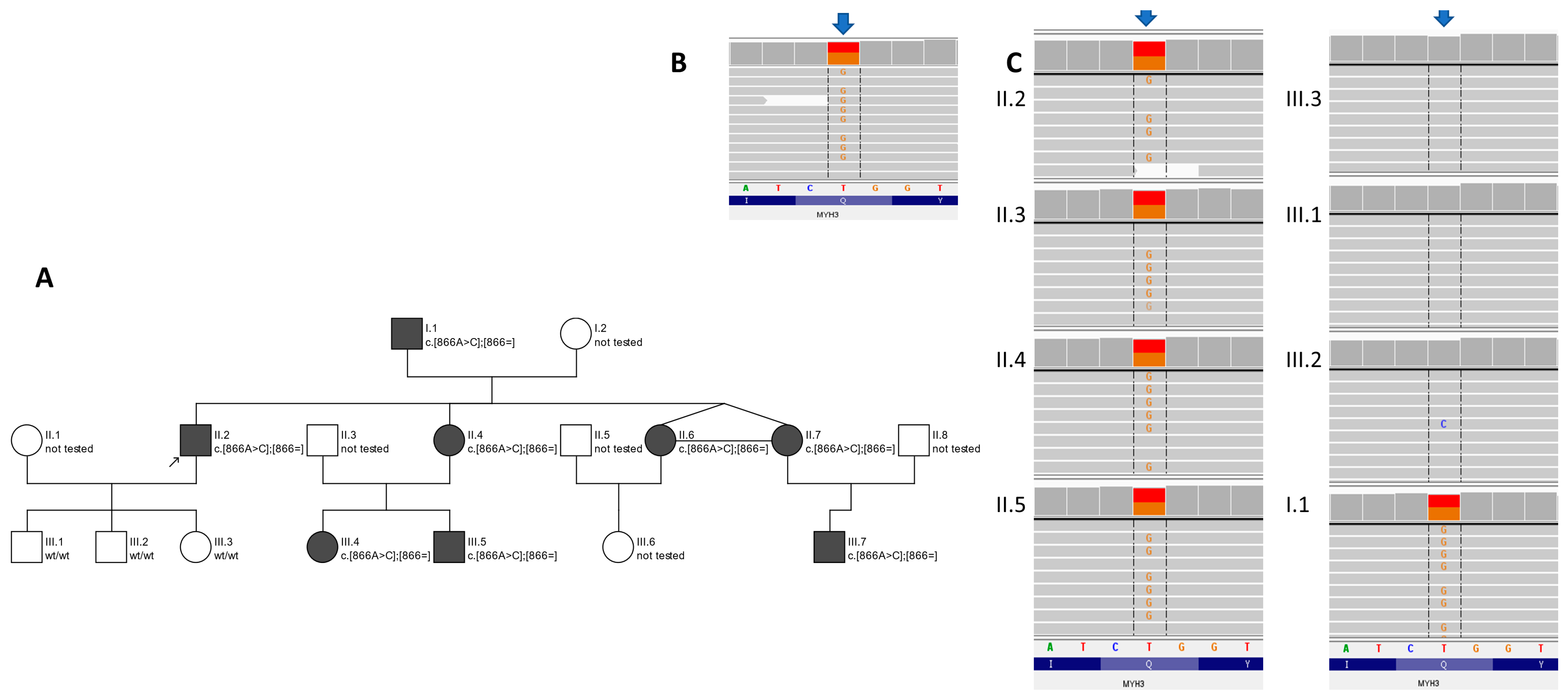
2.1. The Proband
2.2. The Proband’s Family
2.3. Genetic Testing
3. Results
3.1. Clinical Features
3.2. Clinical Characteristics of Patients
- -
- Bony blocks or partial bony blocks between the occipitocervical junctions; the C1 vertebra and C2 vertebra; the L4 vertebra; the L5 vertebra; and the lumcosacral junctions (all subjects);
- -
- Unconjoined/hypoplasia of the posterior arch of the C1 vertebra (subjects II.4 and II.7);
- -
- Spina bifida of the L5 and S1 vertebra (subject II.4);
- -
- Narrow vertebra canal (subjects I.1. and II.2);
- -
- Lumbar vertebrae with atypical morphology (all subjects);
- -
- Congenital lower hypoplastic intervertebral discs (all subjects).
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Z.; Yu, X.; Shen, J. Environmental aspects of congenital scoliosis. Environ. Sci. Pollut. Res. Int. 2015, 22, 5751–5755. [Google Scholar] [CrossRef] [PubMed]
- Passias, P.G.; Poorman, G.W.; Jalai, C.M.; Diebo, B.G.; Vira, S.; Horn, S.R.; Baker, J.F.; Shenoy, K.; Hasan, S.; Buza, J.; et al. Incidence of Congenital Spinal Abnormalities Among Pediatric Patients and Their Association with Scoliosis and Systemic Anomalies. J. Pediatr. Orthop. 2019, 39, e608–e613. [Google Scholar] [CrossRef] [PubMed]
- Toydemir, R.M.; Rutherford, A.; Whitby, F.G.; Jorde, L.B.; Carey, J.C.; Bamshad, M.J. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat. Genet. 2006, 38, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Zieba, J.; Zhang, W.; Chong, J.X.; Forlenza, K.N.; Martin, J.H.; Heard, K.; Grange, D.K.; Butler, M.G.; Kleefstra, T.; Lachman, R.S.; et al. A postnatal role for embryonic myosin revealed by MYH3 mutations that alter TGFbeta signaling and cause autosomal dominant spondylocarpotarsal synostosis. Sci. Rep. 2017, 7, 41803. [Google Scholar] [CrossRef] [PubMed]
- Eller, M.; Stedman, H.H.; Sylvester, J.E.; Fertels, S.H.; Wu, Q.L.; Raychowdhury, M.K.; Rubinstein, N.A.; Kelly, A.M.; Sarkar, S. Human embryonic myosin heavy chain cDNA. Interspecies sequence conservation of the myosin rod, chromosomal locus and isoform specific transcription of the gene. FEBS Lett. 1989, 256, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Karsch-Mizrachi, I.; Travis, M.; Blau, H.; Leinwand, L.A. Expression and DNA sequence analysis of a human embryonic skeletal muscle myosin heavy chain gene. Nucleic. Acids Res. 1989, 17, 6167–6179. [Google Scholar] [CrossRef] [PubMed]
- Rydzanicz, M.; Wachowska, M.; Cook, E.C.; Lisowski, P.; Kuzniewska, B.; Szymanska, K.; Diecke, S.; Prigione, A.; Szczaluba, K.; Szybinska, A.; et al. Novel calcineurin A (PPP3CA) variant associated with epilepsy, constitutive enzyme activation and downregulation of protein expression. Eur. J. Hum. Genet. 2019, 27, 61–69. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V.; et al. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Meerschaut, I.; De Coninck, S.; Steyaert, W.; Barnicoat, A.; Bayat, A.; Benedicenti, F.; Berland, S.; Blair, E.M.; Breckpot, J.; de Burca, A.; et al. A clinical scoring system for congenital contractural arachnodactyly. Genet. Med. 2020, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Vrtovec, T.; Pernus, F.; Likar, B. A review of methods for quantitative evaluation of spinal curvature. Eur. Spine J. 2009, 18, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.; Van Heest, A.E.; Pleasure, D. Arthrogryposis: A review and update. J. Bone Joint. Surg. Am. 2009, 91, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Cameron-Christie, S.R.; Wells, C.F.; Simon, M.; Wessels, M.; Tang, C.Z.N.; Wei, W.; Takei, R.; Aarts-Tesselaar, C.; Sandaradura, S.; Sillence, D.O.; et al. Recessive Spondylocarpotarsal Synostosis Syndrome Due to Compound Heterozygosity for Variants in MYH3. Am. J. Hum. Genet. 2018, 102, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Carapito, R.; Goldenberg, A.; Paul, N.; Pichot, A.; David, A.; Hamel, A.; Dumant-Forest, C.; Leroux, J.; Ory, B.; Isidor, B.; et al. Protein-altering MYH3 variants are associated with a spectrum of phenotypes extending to spondylocarpotarsal synostosis syndrome. Eur. J. Hum. Genet. 2016, 24, 1746–1751. [Google Scholar] [CrossRef] [PubMed]
- Tajsharghi, H.; Kimber, E.; Kroksmark, A.K.; Jerre, R.; Tulinius, M.; Oldfors, A. Embryonic myosin heavy-chain mutations cause distal arthrogryposis and developmental myosin myopathy that persists postnatally. Arch. Neurol. 2008, 65, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kang, Q.L.; Zhang, Z.L. A MYH3 mutation identified for the first time in a Chinese family with Sheldon-Hall syndrome (DA2B). Neuromuscul. Disord. 2018, 28, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Tuncbilek, E.; Alanay, Y. Congenital contractural arachnodactyly (Beals syndrome). Orphanet. J. Rare Dis. 2006, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.; Chen, W.; Li, W.; Wang, S.; Wang, L.; Zhao, Y.; Lin, M.; Ye, Y.; Lin, J.; et al. Diagnostic yield and clinical impact of exome sequencing in early-onset scoliosis (EOS). J. Med. Genet. 2021, 58, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Vivante, A.; Ityel, H.; Pode-Shakked, B.; Chen, J.; Shril, S.; van der Ven, A.T.; Mann, N.; Schmidt, J.M.; Segel, R.; Aran, A.; et al. Exome sequencing in Jewish and Arab patients with rhabdomyolysis reveals single-gene etiology in 43% of cases. Pediatr. Nephrol. 2017, 32, 2273–2282. [Google Scholar] [CrossRef] [PubMed]
- Scala, M.; Accogli, A.; De Grandis, E.; Allegri, A.; Bagowski, C.P.; Shoukier, M.; Maghnie, M.; Capra, V. A novel pathogenic MYH3 mutation in a child with Sheldon-Hall syndrome and vertebral fusions. Am. J. Med. Genet. A 2018, 176, 663–667. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Subject | I.1 | II.2 | II.4 | II.6 | II.7 | III.4 | III.5 | III.7 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Biological attributes | sex | Male | Male | Female | Female | Female | Female | Male | Female | |
| age (years) | 69 | 41 | 38 | 31 | 31 | 13 | 6 | 2 | ||
| Stature and trunk | Physical finding | short stature | + | + | + | + | + | −25c | 25c | 75c |
| short neck | + | − | + | + | + | + | + | − | ||
| Radiological finding | scoliosis | + | + | + | + | + | − | mild | mild | |
| vertebral fusion | + | + | + | − | + | + | − | ? | ||
| Face | Physical finding | downslating palpebral fissures | + | + | + | + | + | + | + | + |
| Upper limb | Physical finding | joint contractures | + | + | + | + | + | − | + | + |
| camptodactyly | + | + | + | + | + | + | − | − | ||
| elongation of the proximal and middle phalanges of the hands | − | + | − | − | − | − | − | − | ||
| Radiological finding | carpal fusion | + | − | − | + | − | ? | ? | ? | |
| Lower limb | Physical finding | dysplasia of the calf muscles | + | + | + | + | + | + | + | − |
| joint contractures | + | + | + | + | + | + | + | mild | ||
| hallux vagus | + | − | + | − | − | + | − | − | ||
| partial syndactyly of the II and III toes | − | + | − | − | − | − | − | − | ||
| Elongation of the proximal phalanges and metatarsal bones | − | + | − | − | − | − | + | − | ||
| Radiological finding | peripheral instability with plantar deviation of the toes at the IP I joints with predominance of the right foot | − | − | + | − | − | + | − | − | |
| tarsal fusion | − | − | − | − | − | ? | ? | ? | ||
| Phenotype | Arthrogryposis, Distal, Type 2A (Freeman–Sheldon) [4] | Arthrogryposis, Distal, Type 2B3 (Sheldon–Hall) [5,6] | Contractures, Pterygia and Spondylocarpotarsal Fusion Syndrome 1A [7,8,9] | Contractures, Pterygia and Spondylocarpotarsal Fusion Syndrome 1B [9] | Family Described within the Presented Study |
|---|---|---|---|---|---|
| inheritance | AD | AD | AD | AR | AD |
| face | very small mouth, pinched lips, H-shaped dimpling of the chin | Triangular face Facial contractures that result in deep nasolabial folds Attached earlobes Downslating palpebral fissures Broad bridge of nose Small mouth | Microcephaly (in some patients) Ptosis cleft palate Downslating palpebral fissures Low-set posteriorly rotted ears Hearing loss (in some patients) | Dysmorphic features Cleft palate (rare) | Downslating palpebral fissures |
| neck | Short neck | n.d. | Short neck Webbed neck | Short neck Webbed neck | Short neck |
| contractures | Multiple (shoulders, elbows, thumbs, hips, knees, toes) | Multiple (shoulders, elbows, fingers, hips, knees, feet) | Elbows, knees, hips (in same patients) | Variable (neck, shoulders, elbows, fingers, hips and/or knees) | Multiple (elbows, hands, fingers, hips, knees, feet) |
| spine | Kyphoscoliosis (frequently develops) Spina bifida occulta | Scoliosis (rare) | Scoliosis Vertebral fusion Hemivertebrae Spondylolisthesis (rare) | Scoliosis Vertebral fusion | Scoliosis |
| hands | Cortical thumbs Camptodactyly Ulnar deviation | Camptodactyly Ulnar deviation Palmar position of the thumb Overlapping finger | Carpal fusion Camptodactyly V finger clinodactyly | Carpal fusion | Camptodactyly and isolated defects, e.g., elongation of the phalanges |
| feet | Talipes equinovarus Contracted toes | Hallux vagus Talipes equinovarus | Tarsal fusion | Tarsal fusion Club foot (rare) | Isolated defects, e.g., hallux vagus, partial syndactyly, peripheral instability with plantar deviation of the toes |
| skin | Skin thickening on fingers | Hypoplastic or absent flexion creases of palms | Variable pterygia (neck, elbows and/or fingers) | Variable pterygia (neck, elbows, fingers and/ or knees) | Abnormal pigmentation Skin dryness |
| hight | Short stature | Short stature | Short stature | n.d. | Short stature |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frasuńska, J.; Pollak, A.; Turczyn, P.; Kutkowska-Kaźmierczak, A.; Pepłowski, J.; Płoski, R.; Tarnacka, B. A Study of Polish Family with Scoliosis and Limb Contractures Expands the MYH3 Disease Spectrum. Genes 2024, 15, 125. https://doi.org/10.3390/genes15010125
Frasuńska J, Pollak A, Turczyn P, Kutkowska-Kaźmierczak A, Pepłowski J, Płoski R, Tarnacka B. A Study of Polish Family with Scoliosis and Limb Contractures Expands the MYH3 Disease Spectrum. Genes. 2024; 15(1):125. https://doi.org/10.3390/genes15010125
Chicago/Turabian StyleFrasuńska, Justyna, Agnieszka Pollak, Paweł Turczyn, Anna Kutkowska-Kaźmierczak, Jakub Pepłowski, Rafał Płoski, and Beata Tarnacka. 2024. "A Study of Polish Family with Scoliosis and Limb Contractures Expands the MYH3 Disease Spectrum" Genes 15, no. 1: 125. https://doi.org/10.3390/genes15010125
APA StyleFrasuńska, J., Pollak, A., Turczyn, P., Kutkowska-Kaźmierczak, A., Pepłowski, J., Płoski, R., & Tarnacka, B. (2024). A Study of Polish Family with Scoliosis and Limb Contractures Expands the MYH3 Disease Spectrum. Genes, 15(1), 125. https://doi.org/10.3390/genes15010125