
The Phylogenetic Relationship of Lamiinae (Coleoptera: Cerambycidae) Using Mitochondrial Genomes


 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Taxon Sampling and Mitochondrial Genome Sequencing
2.2. Mitochondrial Genome Annotation and Sequence Analyses
2.3. Phylogenetic Analyses
3. Results
3.1. General Characteristics of 11 New Mitochondrial Genomes
3.2. A+T-Rich Region
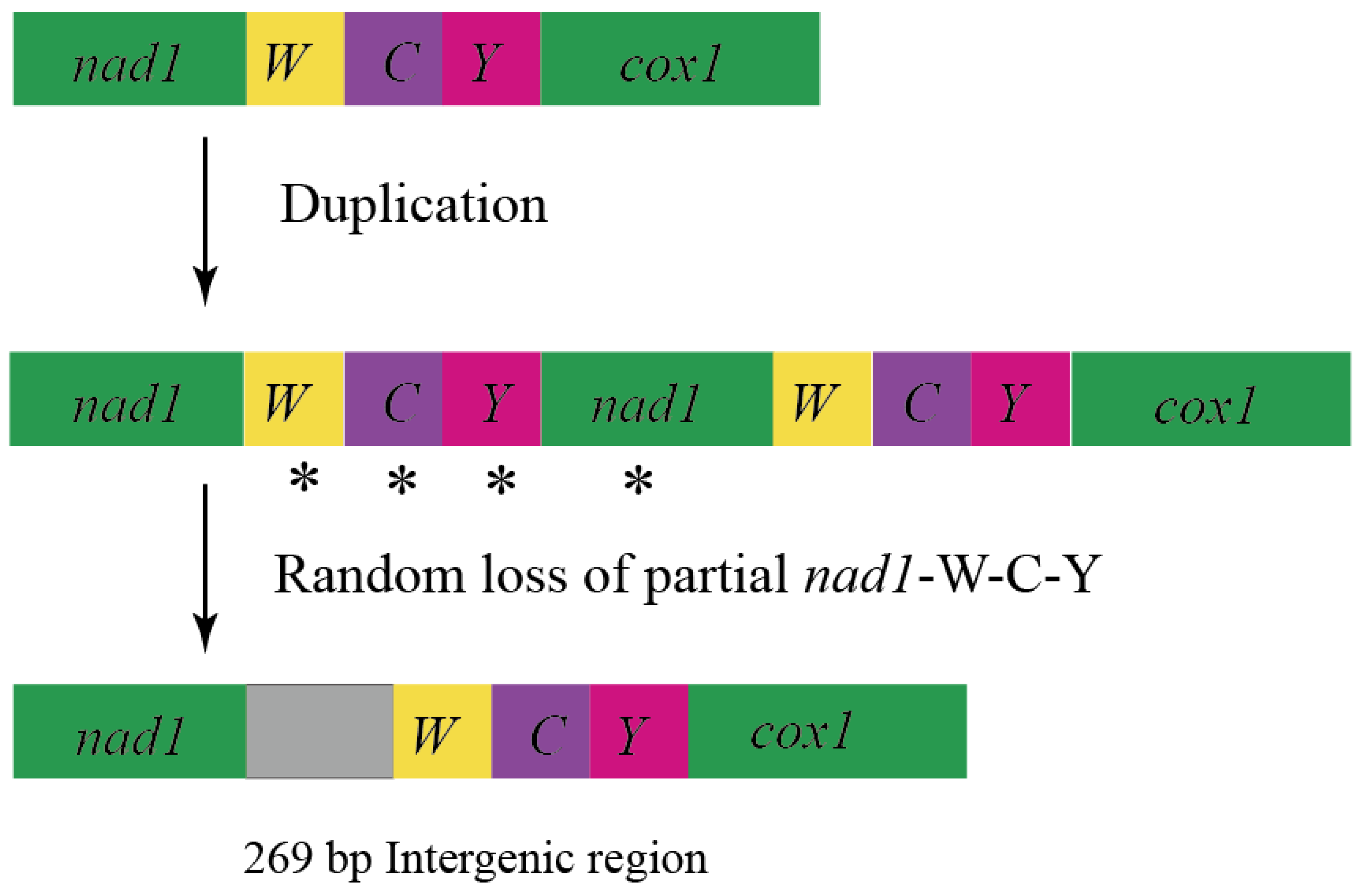
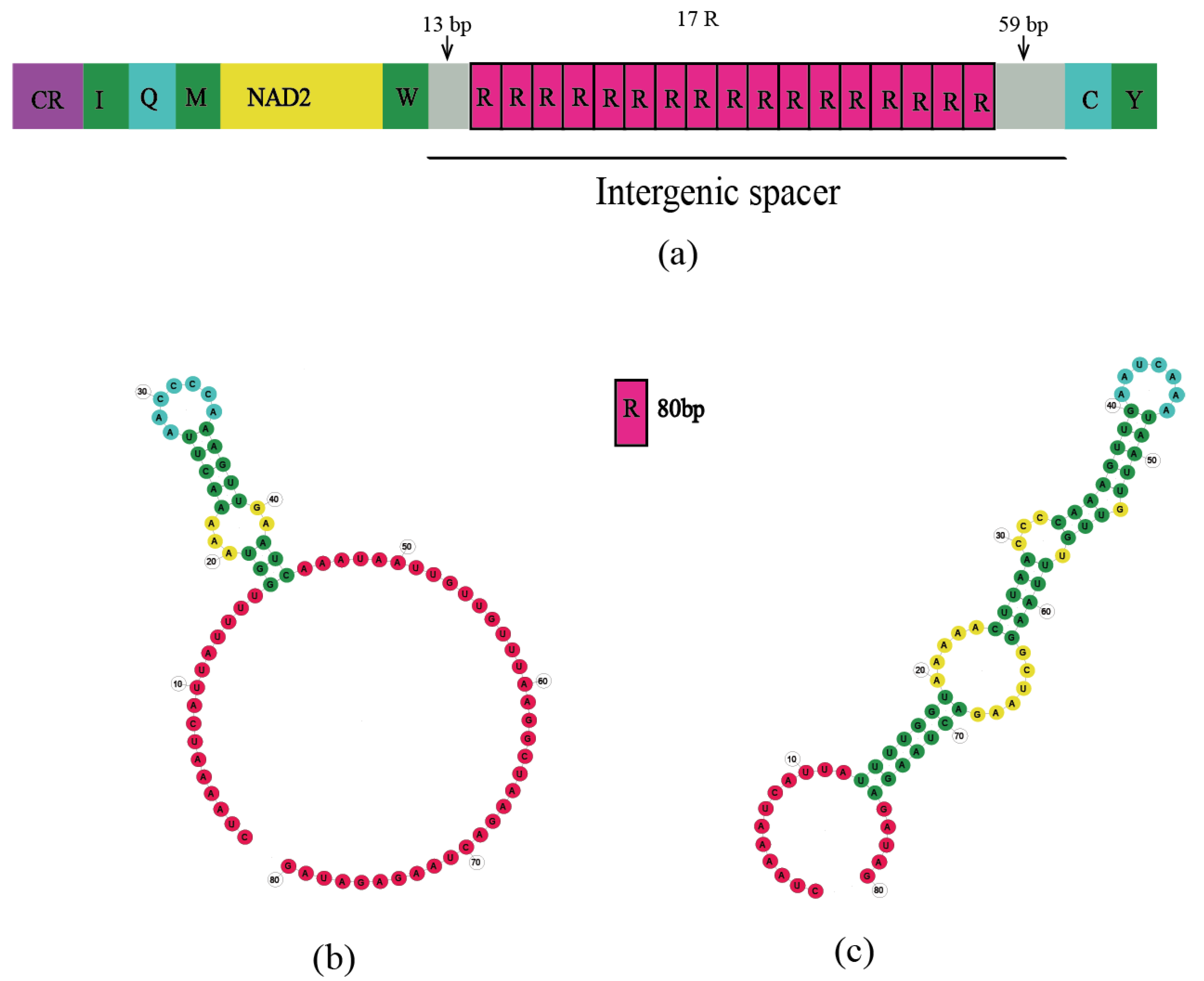
3.3. Intergenic Spacers
3.4. Phylogenetic Analyses
4. Discussion
4.1. General Features of Mitochondrial Genomes
4.2. A+T-Rich Region
4.3. Intergenic Spacers
4.3.1. Short IGS
4.3.2. Long IGS of Mesosa myops
4.3.3. Long IGS of Batocera sp.
4.4. Phylogenetic Analyses
4.4.1. Phylogenetic Analyses of Subfamily
4.4.2. Tribal Classification in Lamiinae
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rossa, R.; Goczał, J. Global diversity and distribution of longhorn beetles (Coleoptera: Cerambycidae). Eur. Zool. J. 2021, 88, 289–302. [Google Scholar] [CrossRef]
- Tavakilian, G.; Chevillotte, H. Titan: Base de données internationales sur les Cerambycidae ou Longicornes. Internet Resour. 2019, 25, 2019. [Google Scholar]
- Wang, B.; Xu, C.; Jarzembowski, E.A. Ecological radiations of insects in the Mesozoic. Trends Ecol. Evol. 2022, 37, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shin, S.; Ashman, L.G.; Leschen, R.A.; Zwick, A.; de Keyzer, R.; McKenna, D.D.; Ślipiński, A. Phylogenomics resolves timing and patterns in the evolution of Australasian Cerambycinae (Coleoptera: Cerambycidae), and reveals new insights into the subfamily-level classification and historical biogeography of longhorn beetles. Mol. Phylogenet. Evol. 2022, 172, 107486. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Ma, J.; McKenna, D.D.; Yan, E.V.; Zhang, H.; Jarzembowski, E.A. The earliest known longhorn beetle (Cerambycidae: Prioninae) and implications for the early evolution of Chrysomeloidea. J. Syst. Palaeontol. 2014, 12, 565–574. [Google Scholar] [CrossRef]
- Lawrence, J.F. Families and subfamilies of coleopteran (with selected genera, notes, references and data on family-group names). In Biology, Phylogeny, and Classification of Coleoptera; Muzeum i Instytut Zoologii PAN: Warszawa, Poland, 1995; pp. 779–1006. ISBN 83-85192-34-4. [Google Scholar]
- Bouchard, P.; Bousquet, Y.; Davies, A.E.; Alonso-Zarazaga, M.A.; Lawrence, J.F.; Lyal, C.H.; Newton, A.F.; Reid, C.A.; Schmitt, M.; Ślipiński, S.A. Family-group names in Coleoptera (Insecta). ZooKeys 2011, 88, 1–972. [Google Scholar] [CrossRef] [PubMed]
- Svacha, P.; Lawrence, J.F. 2.4. Cerambycidae Latreille, 1802. In Volume 3 Morphology and Systematics; De Gruyter: Berlin, Germany, 2014; pp. 77–177. [Google Scholar] [CrossRef]
- Leschen, R.A.; Beutel, R.G. Arthropoda: Insecta: Coleoptera: Volume 3: Morphology and Systematics (Phytophaga); De Gruyter: Berlin, Germany, 2014. [Google Scholar] [CrossRef]
- Shi, F.; Yu, T.; Xu, Y.; Zhang, S.; Niu, Y.; Ge, S.; Tao, J.; Zong, S. Comparative mitochondrial genomic analysis provides new insights into the evolution of the subfamily Lamiinae (Coleoptera: Cerambycidae). Int. J. Biol. Macromol. 2023, 225, 634–647. [Google Scholar] [CrossRef]
- Nie, R.; Vogler, A.P.; Yang, X.K.; Lin, M. Higher-level phylogeny of longhorn beetles (Coleoptera: Chrysomeloidea) inferred from mitochondrial genomes. Syst. Entomol. 2021, 46, 56–70. [Google Scholar] [CrossRef]
- Monné, M.; Wang, Q. General Morphology, Classification and Biology of Cerambycidae; CRC Press: Boca Raton, FL, USA, 2017; pp. 1–70. [Google Scholar]
- Linsley, E.G. Ecology of cerambycidae. Annu. Rev. Entomol. 1959, 4, 99–138. [Google Scholar] [CrossRef]
- Lin, M.Y. A Photographic Guide to Longhorn Beetles of China; Chongqing University Publishing House: Chongqing, China, 2015; pp. 1–226. [Google Scholar]
- Lin, M.Y. Album of Type Specimens of Longhorn Beetles Deposited in National Zoological Museum of China; Henan Science and Technology Press: Henan, China, 2015; pp. 1–374. [Google Scholar]
- Chiang, S.; Pu, F.; Hua, L. Economic Insect Fauna of China, 35, Coleoptera: Cerambycidae (III); Science Press: Beijing China, 1985; pp. 1–189. [Google Scholar]
- de Santana Souza, D.; Marinoni, L.; Monné, M.L.; Gómez-Zurita, J. Molecular phylogenetic assessment of the tribal classification of Lamiinae (Coleoptera: Cerambycidae). Mol. Phylogenet. Evol. 2020, 145, 106736. [Google Scholar] [CrossRef]
- Ashman, L.G.; Shin, S.; Zwick, A.; Ślipiński, A.; McKenna, D.D. The first phylogeny of Australasian Lamiinae longhorn beetles (Coleoptera: Cerambycidae) reveals poor tribal classification and a complex biogeographic history. Syst. Entomol. 2022, 47, 213–230. [Google Scholar] [CrossRef]
- Napp, D. Phylogenetic Relationships among the Subfamilies of Cerambycidae (Coleoptera, Chrysomeloidea); Sociedade Brasileira De Entomologia: São Paulo, Brazil, 1994; pp. 265–419. [Google Scholar]
- Raje, K.R.; Ferris, V.R.; Holland, J.D. Phylogenetic signal and potential for invasiveness. Agric. For. Entomol. 2016, 18, 260–269. [Google Scholar] [CrossRef]
- Haddad, S.; Shin, S.; Lemmon, A.R.; Lemmon, E.M.; Svacha, P.; Farrell, B.; Ślipiński, A.; Windsor, D.; McKenna, D.D. Anchored hybrid enrichment provides new insights into the phylogeny and evolution of longhorned beetles (Cerambycidae). Syst. Entomol. 2018, 43, 68–89. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal Mitochondrial Genomes; Oxford University Press: Oxford, UK, 1999; pp. 1767–1780. [Google Scholar]
- Moritz, C.; Dowling, T.; Brown, W. Evolution of animal mitochondrial DNA: Relevance for population biology and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 269–292. [Google Scholar] [CrossRef]
- Brown, W.M.; George, M., Jr.; Wilson, A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1967–1971. [Google Scholar] [CrossRef] [PubMed]
- Ayivi, S.P.G.; Tong, Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. The mitochondrial genomes of 18 new Pleurosticti (Coleoptera: Scarabaeidae) exhibit a novel trnQ-NCR-trnI-trnM gene rearrangement and clarify phylogenetic relationships of subfamilies within Scarabaeidae. Insects 2021, 12, 1025. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.N.; Yu, P.P.; Zhang, L.P.; Storey, K.B.; Gao, X.Y.; Zhang, J.Y. Increasing 28 mitogenomes of Ephemeroptera, Odonata and Plecoptera support the Chiastomyaria hypothesis with three different outgroup combinations. PeerJ 2021, 9, e11402. [Google Scholar] [CrossRef]
- Zhang, X.; Kang, Z.; Ding, S.; Wang, Y.; Borkent, C.; Saigusa, T.; Yang, D. Mitochondrial genomes provide insights into the phylogeny of Culicomorpha (Insecta: Diptera). Int. J. Mol. Sci. 2019, 20, 747. [Google Scholar] [CrossRef]
- Lin, Y.J.; Zhang, L.H.; Ma, Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Novel gene rearrangements in mitochondrial genomes of four families of praying mantises (Insecta, Mantodea) and phylogenetic relationships of Mantodea. Gene 2023, 880, 147603. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, L.P.; Lin, Y.J.; Yu, D.N.; Storey, K.B.; Zhang, J.Y. Phylogenetic relationships and divergence dating of Mantodea using mitochondrial phylogenomics. Syst. Entomol. 2023, 48, 644–657. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhang, L.; Li, K.; Hong, Y.; Storey, K.B.; Zhang, J.; Yu, D. Nine Mitochondrial Genomes of Phasmatodea with Two Novel Mitochondrial Gene Rearrangements and Phylogeny. Insects 2023, 14, 485. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.K.; Chen, Q.P.; Ayivi, S.P.G.; Guan, J.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Three complete mitochondrial genomes of Orestes guangxiensis, Peruphasma schultei, and Phryganistria guangxiensis (Insecta: Phasmatodea) and their phylogeny. Insects 2021, 12, 779. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; He, K.; Yu, P.; Yu, D.; Cheng, X.; Zhang, J. The complete mitochondrial genomes of three bristletails (Insecta: Archaeognatha): The paraphyly of Machilidae and insights into Archaeognathan phylogeny. PLoS ONE 2015, 10, e0117669. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Shen, C.Y.; Zhao, Y.Y.; Lin, Y.J.; Wu, L.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. The genetic diversity and the divergence time in extant primitive mayfly, Siphluriscus chinensis Ulmer, 1920 using the mitochondrial genome. Genes 2022, 13, 1780. [Google Scholar] [CrossRef] [PubMed]
- Linard, B.; Crampton-Platt, A.; Moriniere, J.; Timmermans, M.J.; Andujar, C.; Arribas, P.; Miller, K.E.; Lipecki, J.; Favreau, E.; Hunter, A. The contribution of mitochondrial metagenomics to large-scale data mining and phylogenetic analysis of Coleoptera. Mol. Phylogenet. Evol. 2018, 128, 1–11. [Google Scholar] [CrossRef]
- Sun, Y.; Sheng, L.; Huang, J.; Zhou, T.; Ma, J.; Wu, S. The complete mitochondrial genome of Batocera lineolata Chevrolat, 1852 (Coleoptera: Cerambycidae). Mitochondrial DNA Part B 2021, 6, 1812–1813. [Google Scholar] [CrossRef]
- Shin, N.R.; Okamura, Y.; Kirsch, R.; Pauchet, Y. Genome sequencing provides insights into the evolution of gene families encoding plant cell wall-degrading enzymes in longhorned beetles. Insect Mol. Biol. 2023, 32, 469–483. [Google Scholar] [CrossRef]
- Bai, Y.; Ye, L.; Yang, K.; Wang, H. Complete mitochondrial genome of Pseudoechthistatus hei (Coleoptera: Cerambycidae: Lamiinae) and its phylogenetic analysis. Mitochondrial DNA Part B 2022, 7, 1997–2001. [Google Scholar] [CrossRef]
- Wang, J.; Dai, X.Y.; Xu, X.D.; Zhang, Z.Y.; Yu, D.N.; Storey, K.B.; Zhang, J.Y. The complete mitochondrial genomes of five longicorn beetles (Coleoptera: Cerambycidae) and phylogenetic relationships within Cerambycidae. PeerJ 2019, 7, e7633. [Google Scholar] [CrossRef]
- Li, F.; Zhang, H.; Wang, W.; Weng, H.; Meng, Z. Complete mitochondrial genome of the Japanese pine sawyer, Monochamus alternatus (Coleoptera: Cerambycidae). Mitochondrial DNA Part A 2016, 27, 1144–1145. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.W.; Liu, J.; Wang, C.H.; Lin, S.K.; Liu, L.X.; Liu, Y.G.; Ji, Y.Q. The complete mitochondrial DNA sequence of Putian Loquat longicorn (Anoplophora chinensis). Mitochondrial DNA Part B 2020, 5, 1147–1148. [Google Scholar] [CrossRef]
- Kim, K.G.; Hong, M.Y.; Kim, M.J.; Im, H.H.; Kim, M.I.; Bae, C.H.; Seo, S.J.; Lee, S.H.; Kim, I. Complete mitochondrial genome sequence of the yellow-spotted long-horned beetle Psacothea hilaris (Coleoptera: Cerambycidae) and phylogenetic analysis among coleopteran insects. Mol. Cells 2009, 27, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Behere, G.; Tay, W.; Firake, D.; Kunz, D.; Burange, P.; Ramamurthy, V. Characterization of draft mitochondrial genome of guava trunk borer, Aristobia reticulator (Fabricius, 1781)(Coleoptera: Cerambycidae: Lamiinae) from India. Mitochondrial DNA Part B 2019, 4, 1592–1593. [Google Scholar] [CrossRef]
- Zhang, B.L.; Zhang, J.; Zhang, D.; Feng, Y.; Qiu, J.; Ye, X.J.; Wang, B.X. Complete mitochondrial genome of the longicorn Anoplophora horsfieldi Hope (Coleoptera: Cerambycidae). Mitochondrial DNA Part B 2023, 8, 220–221. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, X.; Qian, L.; An, Y.; Fang, J. The complete mitochondrial genome of the citrus long-horned beetle, Anoplophora chinensis (Coleoptera: Cerambycidae). Mitochondrial DNA Part A 2016, 27, 4665–4667. [Google Scholar] [CrossRef]
- Dai, X.Y.; Zhang, H.; Xu, X.D.; Jia, Y.Y.; Zhang, J.Y.; Yu, D.N.; Cheng, H.Y. The complete mitochondrial genome of Annamanum lunulatum (Coleoptera: Lamiinae) and its phylogeny. Mitochondrial DNA Part B 2020, 5, 551–553. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Guan, J.Y.; Cao, Y.R.; Dai, X.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Mitogenome analysis of four Lamiinae species (Coleoptera: Cerambycidae) and gene expression responses by Monochamus alternatus when infected with the parasitic nematode, Bursaphelenchus mucronatus. Insects 2021, 12, 453. [Google Scholar] [CrossRef]
- Liu, J.H.; Jia, P.F.; Luo, T.; Wang, Q.M. Complete mitochondrial genome of white-striped long-horned beetle, Batocera lineolata (Coleoptera: Cerambycidae) by next-generation sequencing and its phylogenetic relationship within superfamily Chrysomeloidea. Mitochondrial DNA Part B 2017, 2, 520–521. [Google Scholar] [CrossRef]
- Wang, C.Y.; Feng, Y.; Chen, X.M. Complete coding region of the mitochondrial genome of Monochamus alternatus Hope (Coleoptera: Cerambycidae). Zool. Sci. 2013, 30, 570–576. [Google Scholar] [CrossRef]
- Crampton-Platt, A.; Timmermans, M.J.; Gimmel, M.L.; Kutty, S.N.; Cockerill, T.D.; Vun Khen, C.; Vogler, A.P. Soup to tree: The phylogeny of beetles inferred by mitochondrial metagenomics of a Bornean rainforest sample. Mol. Biol. Evol. 2015, 32, 2302–2316. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Lu, H.; Chen, L.; Sabatelli, S.; Wang, C.; Xie, G.; Wang, P.; Liu, M.; Wang, W.; Audisio, P. Comparative mitogenomic analysis of two longhorn beetles (Coleoptera: Cerambycidae: Lamiinae) with preliminary investigation into phylogenetic relationships of tribes of Lamiinae. Insects 2021, 12, 820. [Google Scholar] [CrossRef] [PubMed]
- de Sena Brandine, G.; Smith, A.D. Falco: High-speed FastQC emulation for quality control of sequencing data. F1000 Res. 2019, 8, 1874. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; DePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Jühling, F.; Pütz, J.; Bernt, M.; Donath, A.; Middendorf, M.; Florentz, C.; Stadler, P.F. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 2012, 40, 2833–2845. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Lalitha, S. Primer premier 5. Biotech Softw. Internet Rep. Comput. Softw. J. Sci. 2000, 1, 270–272. [Google Scholar] [CrossRef]
- Sanger, F. Determination of nucleotide sequences in DNA. Science 1981, 214, 1205–1210. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Stothard, P.; Grant, J.R.; Van Domselaar, G. Visualizing and comparing circular genomes using the CGView family of tools. Brief. Bioinform. 2019, 20, 1576–1582. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Johnson, S. Adobe Illustrator CS5 on Demand; Que Publishing: Hoboken, NJ, USA, 2010; pp. 1–508. ISBN -100789744457. [Google Scholar]
- Xia, X.; Xie, Z. DAMBE: Software package for data analysis in molecular biology and evolution. J. Hered. 2001, 92, 371–373. [Google Scholar] [CrossRef]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L.; Lavrov, D.V.; Brown, W.M. Gene translocation links insects and crustaceans. Nature 1998, 392, 667–668. [Google Scholar] [CrossRef]
- Song, N.; Liu, H.Y.; Yang, X.J.; Zhao, X.C.; Lin, A.L. Complete mitochondrial genome of the darkling beetle Gonocephalum outreyi (Coleoptera: Tenebrionidae) with phylogenetic implications. J. Asia-Pac. Entomol. 2018, 21, 721–730. [Google Scholar] [CrossRef]
- Friedrich, M.; Muqim, N. Sequence and phylogenetic analysis of the complete mitochondrial genome of the flour beetle Tribolium castanaeum. Mol. Phylogenet. Evol. 2003, 26, 502–512. [Google Scholar] [CrossRef]
- Bae, J.S.; Kim, I.; Sohn, H.D.; Jin, B.R. The mitochondrial genome of the firefly, Pyrocoelia rufa: Complete DNA sequence, genome organization, and phylogenetic analysis with other insects. Mol. Phylogenet. Evol. 2004, 32, 978–985. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Donath, A.; Jühling, F.; Al-Arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef]
- Jank, P.; Riesner, D.; Gross, H.J. Rabbit liver tRNA1Val: II. Unusual secondary structure of TΨC stem and loop due to a U54: A60 base pair. Nucleic Acids Res. 1977, 4, 2009–2020. [Google Scholar] [CrossRef]
- Yamazaki, N.; Ueshima, R.; Terrett, J.A.; Yokobori, S.-i.; Kaifu, M.; Segawa, R.; Kobayashi, T.; Numachi, K.-i.; Ueda, T.; Nishikawa, K. Evolution of pulmonate gastropod mitochondrial genomes: Comparisons of gene organizations of Euhadra, Cepaea and Albinaria and implications of unusual tRNA secondary structures. Genetics 1997, 145, 749–758. [Google Scholar] [CrossRef]
- Hanada, T.; Suzuki, T.; Yokogawa, T.; Takemotootoi, S.; Kaifu, M.; Segawa, R.; Kobayashi, T. Numachi, mitochondrial tRNAs Ser with unusual secondary structures in an in vitro translation system of bovine mitochondria. Genes Cells 2001, 6, 1019–1030. [Google Scholar] [CrossRef]
- Negrisolo, E.; Babbucci, M.; Patarnello, T. The mitochondrial genome of the ascalaphid owlfly Libelloides macaronius and comparative evolutionary mitochondriomics of neuropterid insects. BMC Genom. 2011, 12, 221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Cameron, S. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research. Syst. Entomol. 2014, 39, 400–411. [Google Scholar] [CrossRef]
- Tørresen, O.K.; Star, B.; Mier, P.; Andrade-Navarro, M.A.; Bateman, A.; Jarnot, P.; Gruca, A.; Grynberg, M.; Kajava, A.V.; Promponas, V.J. Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res. 2019, 47, 10994–11006. [Google Scholar] [CrossRef]
- Buroker, N.; Brown, J.; Gilbert, T.; O’hara, P.; Beckenbach, A.; Thomas, W.; Smith, M. Length heteroplasmy of sturgeon mitochondrial DNA: An illegitimate elongation model. Genetics 1990, 124, 157–163. [Google Scholar] [CrossRef]
- Mignotte, F.; Gueride, M.; Champagne, A.M.; Mounolou, J.C. Direct repeats in the non-coding region of rabbit mitochondrial DNA: Involvement in the generation of intra-and inter-individual heterogeneity. Eur. J. Biochem. 1990, 194, 561–571. [Google Scholar] [CrossRef]
- Saito, S.; Tamura, K.; Aotsuka, T. Replication origin of mitochondrial DNA in insects. Genetics 2005, 171, 1695–1705. [Google Scholar] [CrossRef]
- Crozier, R.; Crozier, Y. The mitochondrial genome of the honeybee Apis mellifera: Complete sequence and genome organization. Genetics 1993, 133, 97–117. [Google Scholar] [CrossRef]
- Franck, P.; Garnery, L.; Solignac, M.; Cornuet, J.M. The origin of west European subspecies of honeybees (Apis mellifera): New insights from microsatellite and mitochondrial data. Evolution 1998, 52, 1119–1134. [Google Scholar] [CrossRef] [PubMed]
- Rodovalho, C.d.M.; Lyra, M.L.; Ferro, M.; Bacci Jr, M. The mitochondrial genome of the leaf-cutter ant Atta laevigata: A mitogenome with a large number of intergenic spacers. PLoS ONE 2014, 9, e97117. [Google Scholar] [CrossRef] [PubMed]
- Dotson, E.; Beard, C. 3 Sequence and organization of the mitochondrial genome of the Chagas disease vector, Triatoma dimidiata. Insect Mol. Biol. 2001, 10, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Kim, S.; Wan, X. Mitochondrial genomes of the Dorcus velutinus complex (Coleoptera: Lucanidae) with the large intergenic spacer showing unique short sequence repeats and their implications for systematics. J. Asia-Pac. Entomol. 2021, 24, 493–501. [Google Scholar] [CrossRef]
- Yuan, M.L.; Zhang, Q.L.; Zhang, L.; Guo, Z.L.; Liu, Y.J.; Shen, Y.Y.; Shao, R. High-level phylogeny of the Coleoptera inferred with mitochondrial genome sequences. Mol. Phylogenet. Evol. 2016, 104, 99–111. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Chen, D.B.; Liu, H.H.; Hu, H.L.; Bian, H.X.; Zhang, R.S.; Yang, R.S.; Jiang, X.F.; Shi, S.L. The complete mitochondrial genome of the longhorn beetle Dorysthenes paradoxus (Coleoptera: Cerambycidae: Prionini) and the implication for the phylogenetic relationships of the Cerambycidae species. J. Insect Sci. 2018, 18, 21. [Google Scholar] [CrossRef]
- Wei, S.J.; Tang, P.; Zheng, L.H.; Shi, M.; Chen, X.X. The complete mitochondrial genome of Evania appendigaster (Hymenoptera: Evaniidae) has low A + T content and a long intergenic spacer between atp8 and atp6. Mol. Biol. Rep. 2010, 37, 1931–1942. [Google Scholar] [CrossRef]
- Dai, L.; Qian, C.; Zhang, C.; Wang, L.; Wei, G.; Li, J.; Zhu, B.; Liu, C. Characterization of the complete mitochondrial genome of Cerura menciana and comparison with other lepidopteran insects. PLoS ONE 2015, 10, e0132951. [Google Scholar] [CrossRef]
- Du, C.; Zhang, L.; Lu, T.; Ma, J.; Zeng, C.; Yue, B.; Zhang, X. Mitochondrial genomes of blister beetles (Coleoptera, Meloidae) and two large intergenic spacers in Hycleus genera. BMC Genom. 2017, 18, 698. [Google Scholar] [CrossRef]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Yan, L.; Hou, Z.; Ma, J.; Wang, H.; Gao, J.; Zeng, C.; Chen, Q.; Yue, B.; Zhang, X. Complete mitochondrial genome of Episymploce splendens (Blattodea: Ectobiidae): A large intergenic spacer and lacking of two tRNA genes. PLoS ONE 2022, 17, e0268064. [Google Scholar] [CrossRef]
- Sheffield, N.; Song, H.; Cameron, S.; Whiting, M. A comparative analysis of mitochondrial genomes in Coleoptera (Arthropoda: Insecta) and genome descriptions of six new beetles. Mol. Biol. Evol. 2008, 25, 2499–2509. [Google Scholar] [CrossRef]
- Wetterer, J.K.; Schultz, T.R.; Meier, R. Phylogeny of fungus-growing ants (tribe Attini) based on mtDNA sequence and morphology. Mol. Phylogenet. Evol. 1998, 9, 42–47. [Google Scholar] [CrossRef]
- Kronauer, D.; Hölldobler, B.; Gadau, J. Phylogenetics of the new world honey ants (genus Myrmecocystus) estimated from mitochondrial DNA sequences. Mol. Phylogenet. Evol. 2004, 32, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Bacci Jr, M.; Solomon, S.E.; Mueller, U.G.; Martins, V.G.; Carvalho, A.O.; Vieira, L.G.; Silva-Pinhati, A.C.O. Phylogeny of leafcutter ants in the genus Atta Fabricius (Formicidae: Attini) based on mitochondrial and nuclear DNA sequences. Mol. Phylogenet. Evol. 2009, 51, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhou, S.; Wan, X. Phylogenetic implication of large intergenic spacers: Insights from a mitogenomic comparison of Prosopocoilus stag beetles (Coleoptera: Lucanidae). Animals 2022, 12, 1595. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kim, K.G.; Kim, S.R.; Kim, I. Complete mitochondrial genome of the two-spotted stag beetle, Metopodontus blanchardi (Coleoptera: Lucanidae). Mitochondrial DNA 2015, 26, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Galileo, M.H.M.; Rosa, P.; Santos-Silva, A. Two new replacements names for South American Cerambycinae (Coleoptera, Cerambycidae). Zootaxa 2020, 4834, zootaxa. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Chen, L.; Yan-Li, E.; Zheng, K.-D. Phylogenetic analysis of the Prionini (Coleoptera: Cerambycidae: Prioninae) from China based on mitochondrial ribosomal RNA genes and Cytochrome oxidase I gene. Zootaxa 2010, 2487, 1–18. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Ren, B. Molecular phylogeny of some longhorned beetles based on 28S rDNA sequences. Sci. Silvae Sin. 2012, 48, 86–94. [Google Scholar] [CrossRef]
- Wei, Z.; Yin, X.; An, S.; Su, L.; Li, J.; Zhang, H. Molecular phylogenetic study of the higher taxa of the superfamily Cerambycoidea (Insecta: Coleoptera) based on the combined analysis of ribosomal DNA sequences. Acta Entomol. Sin. 2014, 57, 710–720. [Google Scholar]
- Lee, S.; Lee, S. Multigene phylogeny uncovers oviposition-related evolutionary history of Cerambycinae (Coleoptera: Cerambycidae). Mol. Phylogenet. Evol. 2020, 145, 106707. [Google Scholar] [CrossRef]
- Haddad, S.; Gutiérrez, N.; Noguera, F.A.; Shin, S.; Svacha, P.; McKenna, D.D. Phylogenetic placement of the enigmatic longhorned beetle Vesperoctenus flohri Bates (Vesperidae) and a first description of its female internal structures. Arthropod Syst. Phylogeny 2021, 79, 99–114. [Google Scholar] [CrossRef]
- Linsley, E.G.; John, A.C. The Cerambycidae of North America; University of California Press: Oakland, CA, USA, 1961; Volume 102, pp. 1–292. [Google Scholar]
- Linsley, E.G. The Cerambycidae of North America: Taxonomy and Classification of the Parandrinae, Prioninae, Spondylinae, and Aseminae; University of California Press: Oakland, CA, USA, 1962; Volume 18, pp. 1–102. [Google Scholar]
- Gressitt, J.L. Cerambycids of Laos (Disteniidae, Prioninae, Philinae, Aseminae, Lepturinae, Cerambycinae). Pac. Insects Monograph. 1970, 24, 1–314. [Google Scholar] [CrossRef]
- Švácha, P.; Danilevsky, M. Cerambycoid Larvae of Europe and Soviet Union. Part 1. Acta Universitatis Carolinae; Biologica, Karolinum Press: Karolinum, Czech Republic, 1987; pp. 1–186. [Google Scholar]
- Obayashi, N.; Ōbayashi, N.; Niisato, T. Longicorn Beetles of Japan; Tokai University Press: Tokyo, Japan, 2007; pp. 1–842. [Google Scholar]
- Löbl, I.; Smetana, A. Catalogue of Palaearctic Coleoptera. Volume 6: Chrysomeloidea; Apollo Books: New York, NY, USA, 2010. [Google Scholar] [CrossRef]
- Lin, M. Insect Fauna of the Qinling Mountains, Volume VI (Coleoptera II), Cerambycid-Beetles; Xi’an World Publishing Corporation: Xi’an, China, 2017; ISBN 9787519240400. [Google Scholar]
- Crowson, R.A. The Biology of the Coleoptera; Academic Press: Cambridge, UK, 2013; pp. 1–789. ISBN 9781483217604. [Google Scholar]
- Nie, R.; Lin, M.; Xue, H.; Bai, M.; Yang, X. Complete mitochondrial genome of Spiniphilus spinicornis (Coleoptera: Vesperidae: Philinae) and phylogenetic analysis among Cerambycoidea. Mitochondrial DNA Part A 2017, 28, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Linsley, E.G. A revision of the north American Necydalini (Coleoptera, Cerambycidae). Ann. Entomol. Soc. Am. 1940, 33, 269–281. [Google Scholar] [CrossRef]
- Gressitt, J.L. Longicorn Beetles of China; Longicornia: Paris, French, 1951; Volume 2, pp. 1–667. [Google Scholar]
- Linsley, E.; Chemsak, J. Cerambycidae of North America. Part VI, No. 2. In Taxonomy and Classification of the Subfamily Lepturinae [New Taxa]; Publications in Entomology-California University: Oakland, CA, USA, 1976; pp. 1–186. [Google Scholar]
- Wang, Q.; Chiang, S. The Evolution in the Higher Taxa of the Cerambycidae (Coleoptera); Entomotaxonomia: Shanxi, China, 1991; pp. 93–114. [Google Scholar]
- Monné, M.A.; Giesbert, E.F. Checklist of the Cerambycidae and Disteniidae (Coleoptera) of the Western Hemisphere; Wolfsgarden Books: Madison, WI, USA, 1995; pp. 1–420. ISBN 9781885850010, 1885850018. [Google Scholar]
- Monne, M.A. Catalogue of the Cerambycidae (Coleoptera) of the Neotropical Region. Part III. Subfamilies Parandrinae, Prioninae, Anoplodermatinae, Aseminae, Spondylidinae, Lepturinae, Oxypeltinae, and addenda to the Cerambycinae and Lamiinae. Zootaxa 2006, 1212, 1–244. [Google Scholar] [CrossRef]
- Nakamura, S. Morphological and Taxonomic Studies of the Cerambycid Pupae of Japan: (Coleoptera: Cerambycidae); Wako Shoji Corporation: Fujisawa Shi, Japan, 1981; pp. 154–159. [Google Scholar]
- Duffy, E.A.J. Monograph of the Immature Stages of British and Imported Timber Beetles (Cerambycidae); British Museum: London, UK, 1953; pp. 1–351. [Google Scholar]
- Sýkorová, M. Molecular Phylogenesis of the Subfamilies Spondylidinae and Lepturinae (Coleoptera: Cerambycidae) Based on Mitochondrial 16S rDNA. Bachelor’s Thesis, Jihočeská Univerzita v Českých Budějovicích Přírodovědecká Fakulta, České Budějovice, Czech Republic, 2008; pp. 1–38. [Google Scholar]
- Gomez-Zurita, J.; Hunt, T.; Kopliku, F.; Vogler, A.P. Recalibrated tree of leaf beetles (Chrysomelidae) indicates independent diversification of angiosperms and their insect herbivores. PLoS ONE 2007, 2, e360. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Zurita, J.; Hunt, T.; Vogler, A.P. Multilocus ribosomal RNA phylogeny of the leaf beetles (Chrysomelidae). Cladistics 2008, 24, 34–50. [Google Scholar] [CrossRef]
- Marvaldi, A.E.; Duckett, C.N.; Kjer, K.M.; Gillespie, J.J. Structural alignment of 18S and 28S rDNA sequences provides insights into phylogeny of Phytophaga (Coleoptera: Curculionoidea and Chrysomeloidea). Zool. Scr. 2009, 38, 63–77. [Google Scholar] [CrossRef]
- Saha, S.; Özdikmen, H.; Biswas, M.K.; Raychaudhuri, D. Exploring flat faced longhorn beetles (Cerambycidae: Lamiinae) from the reserve forests of Dooars, West Bengal, India. Int. Sch. Res. Not. 2013, 2013, 737193. [Google Scholar] [CrossRef]
- Toki, W.; Kubota, K. Molecular phylogeny based on mitochondrial genes and evolution of host plant use in the long-horned beetle tribe Lamiini (Coleoptera: Cerambycidae) in Japan. Environ. Entomol. 2010, 39, 1336–1343. [Google Scholar] [CrossRef]
- Sama, G. Preliminary note on the Cerambycid fauna of north Africa with the description of new taxa. Quad. Studi E Not. Stor. Nat. Della Romagna 2008, 27, 217–245. [Google Scholar]
- Bi, W.X.; Lin, M.Y. A revision of the genus Pseudoechthistatus Pic (Coleoptera, Cerambycidae, Lamiinae, Lamiini). ZooKeys 2016, 604, 49–85. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Subfamily |
|---|---|
| Lawrence and Newton [6] | Anoplodermatinae, Apatophyseinae, Cerambycinae, Disteniinae, Oxypeltinae, Lepturinae, Lamiinae, Necydalinae, Parandrinae, Philinae, Prioninae, Spondylidinae, Vesperinae |
| Bouchard et al. [7] | Apatophyseinae, Cerambycinae, Dorcasominae, Lamiinae, Lepturinae, Necydalinae, Parandrinae, Prioninae, Spondylidinae |
| Svacha and Lawrence [8] | Cerambycinae, Dorcasominae, Lepturinae, Lamiinae, Necydalinae, Parandrinae, Prioninae, Spondylidinae |
| Species | Accession No. | Length (bp) | Tribe |
|---|---|---|---|
| Acalolepta permutans | OR149089 | 15,500 | Lamiini |
| Batocera sp. | OR149086 | 16,843 | Batocerini |
| Eutetrapha metallescens | OR149087 | 15,505 | Saperdini |
| Glenea pulchra | OR149088 | 15,470 | Saperdini |
| Lamiomimus gottschei | OR149090 | 16,421 | Lamiini |
| Macrochenus isabellinus | OR149091 | 15,675 | Lamiini |
| Mesosa myops | OR149092 | 18,499 | Mesosini |
| Oberea vittata | OR149093 | 15,494 | Saperdini |
| Pharsalia subgemmata | OR149094 | 16,553 | Lamiini |
| Rondibilis sp. | OR149095 | 15,854 | Acanthocinini |
| Saperda subobliterata | OR149096 | 15,499 | Saperdini |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, K.; Yu, S.-W.; Hu, H.; Feng, Y.-F.; Storey, K.B.; Ma, Y.; Zhang, J.-Y.; Yu, D.-N. The Phylogenetic Relationship of Lamiinae (Coleoptera: Cerambycidae) Using Mitochondrial Genomes. Genes 2024, 15, 13. https://doi.org/10.3390/genes15010013
Li K, Yu S-W, Hu H, Feng Y-F, Storey KB, Ma Y, Zhang J-Y, Yu D-N. The Phylogenetic Relationship of Lamiinae (Coleoptera: Cerambycidae) Using Mitochondrial Genomes. Genes. 2024; 15(1):13. https://doi.org/10.3390/genes15010013
Chicago/Turabian StyleLi, Ke, Sheng-Wu Yu, Hao Hu, Yu-Feng Feng, Kenneth B. Storey, Yue Ma, Jia-Yong Zhang, and Dan-Na Yu. 2024. "The Phylogenetic Relationship of Lamiinae (Coleoptera: Cerambycidae) Using Mitochondrial Genomes" Genes 15, no. 1: 13. https://doi.org/10.3390/genes15010013
APA StyleLi, K., Yu, S.-W., Hu, H., Feng, Y.-F., Storey, K. B., Ma, Y., Zhang, J.-Y., & Yu, D.-N. (2024). The Phylogenetic Relationship of Lamiinae (Coleoptera: Cerambycidae) Using Mitochondrial Genomes. Genes, 15(1), 13. https://doi.org/10.3390/genes15010013