

Novel Homozygous FA2H Variant Causing the Full Spectrum of Fatty Acid Hydroxylase-Associated Neurodegeneration (SPG35)

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genetics
2.1.1. High Throughput Sequencing and Bioinformatics Pipeline
2.1.2. Nomenclature, Interpretation, and Classification of Genetic Variants
2.2. Structural Modeling of the Cys25Trp Variant
2.3. Scanning Electron Microscopy of Hair Shafts
3. Results
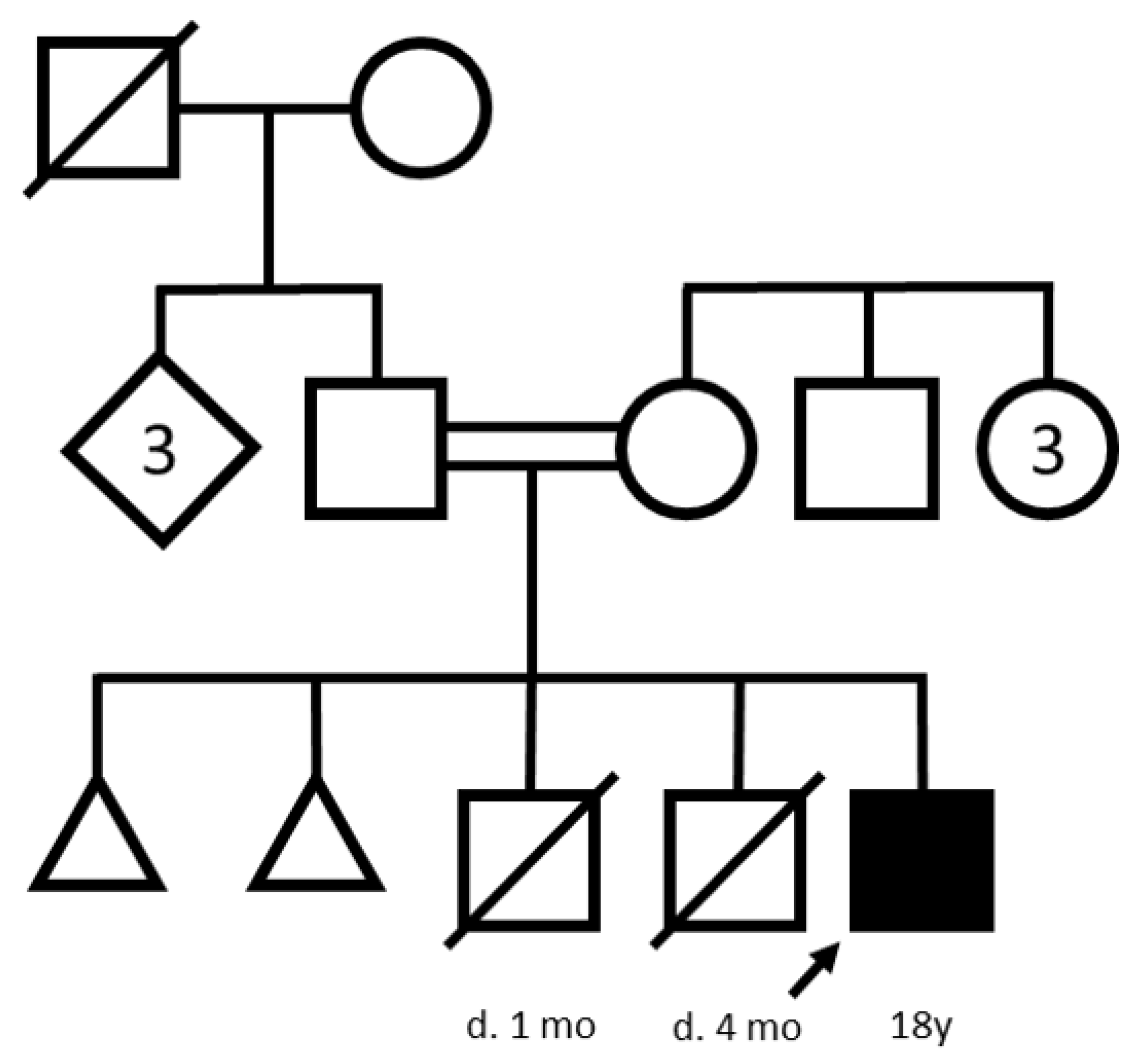
3.1. Case Report
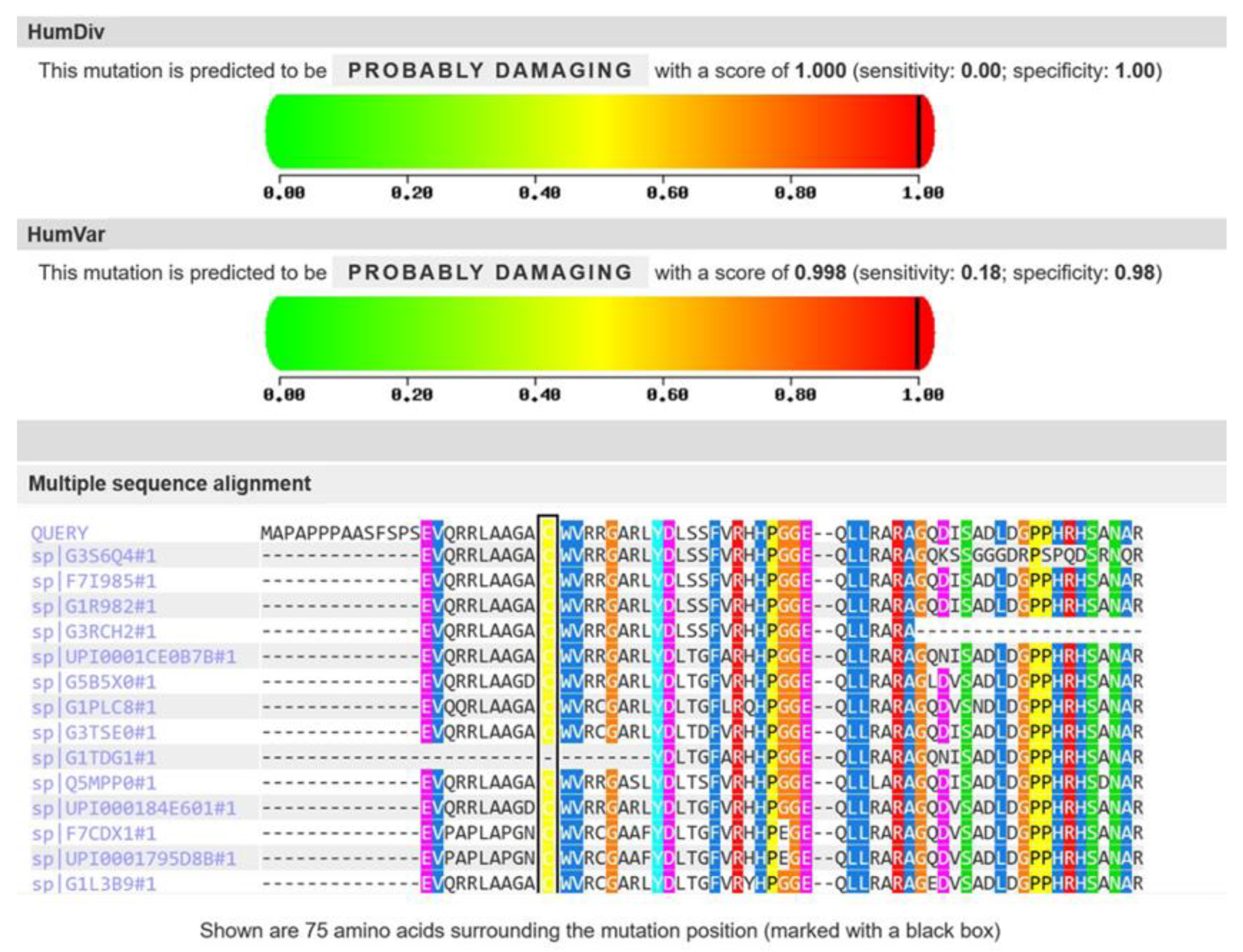
3.2. Molecular Genetic Analysis
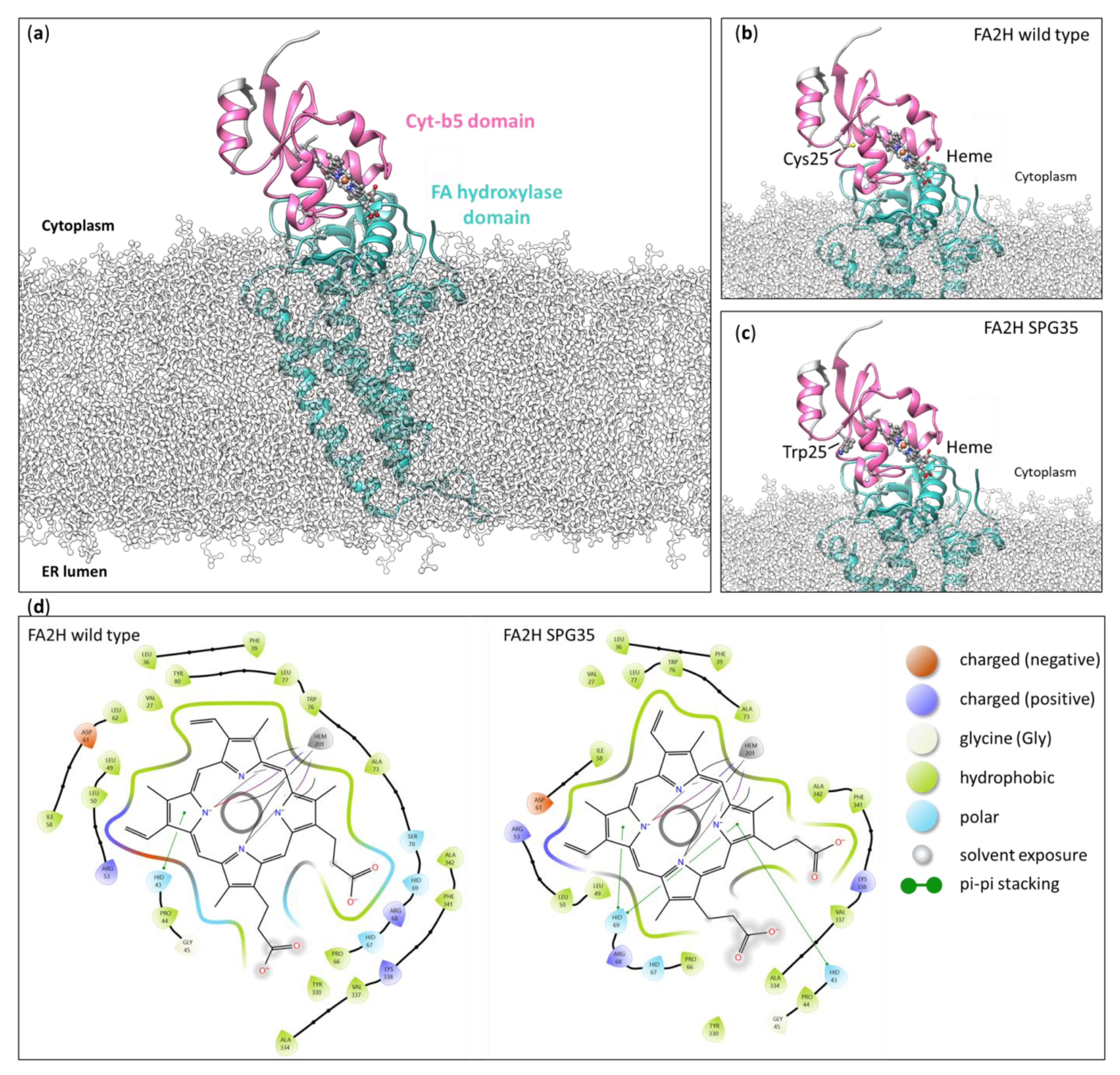
3.3. Structural Modeling of the Cys25Trp Variant
3.4. Scanning Electron Microscopy of Hair Shafts
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fink, J.K. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013, 126, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Rattay, T.W.; Lindig, T.; Baets, J.; Smets, K.; Deconinck, T.; Söhn, A.S.; Hörtnagel, K.; Eckstein, K.N.; Wiethoff, S.; Reichbauer, J.; et al. FAHN/SPG35: A narrow phenotypic spectrum across disease classifications. Brain 2019, 142, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Dan, P.; Edvardson, S.; Bielawski, J.; Hama, H.; Saada, A. 2-Hydroxylated sphingomyelin profiles in cells from patients with mutated fatty acid 2-hydroxylase. Lipids Health Dis. 2011, 10, 84. [Google Scholar] [CrossRef]
- Kishimoto, Y.; Radin, N.S. Occurrence of 2-hydroxy fatty acids in animal tissues. J. Lipid Res. 1963, 4, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Kanfer, J.N. Ceramide galactoside of enriched neuronal and glial fractions from rat brain. J. Biol. Chem. 1972, 247, 1055–1056. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zhou, D.; Pryse, K.M.; Okunade, A.L.; Su, X. Fatty acid 2-hydroxylase mediates diffusional mobility of Raft-associated lipids, GLUT4 level, and lipogenesis in 3T3-L1 adipocytes. J. Biol. Chem. 2010, 285, 25438–25447. [Google Scholar] [CrossRef]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Mari, F.; Berti, B.; Romano, A.; Baldacci, J.; Rizzi, R.; Grazia Alessandrì, M.; Tessa, A.; Procopio, E.; Rubegni, A.; Lourenḉo, C.M.; et al. Clinical and neuroimaging features of autosomal recessive spastic paraplegia 35 (SPG35): Case reports, new mutations, and brief literature review. Neurogenetics 2018, 19, 123–130. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Altuve, A.; Terzyan, S.; Zhang, X.; Kuczera, K.; Rivera, M.; Benson, D.R. Accommodating a nonconservative internal mutation by water-mediated hydrogen bonding between β-sheet strands: A comparison of human and rat type B (mitochondrial) cytochrome b5. Biochemistry 2011, 50, 5544–5554. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Koszelak-Rosenblum, M.; Connelly, S.M.; Dumont, M.E.; Malkowski, M.G. The Crystal Structure of an Integral Membrane Fatty Acid α-Hydroxylase. J. Biol. Chem. 2015, 290, 29820–29833. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Weitkamp, J.T.; Wöltje, M.; Nußpickel, B.; Schmidt, F.N.; Aibibu, D.; Bayer, A.; Eglin, D.; Armiento, A.R.; Arnold, P.; Cherif, C.; et al. Silk Fiber-Reinforced Hyaluronic Acid-Based Hydrogel for Cartilage Tissue Engineering. Int. J. Mol. Sci. 2021, 22, 3635. [Google Scholar] [CrossRef] [PubMed]
- Cabron, A.S.; El Azzouzi, K.; Boss, M.; Arnold, P.; Schwarz, J.; Rosas, M.; Dobert, J.P.; Pavlenko, E.; Schumacher, N.; Renné, T.; et al. Structural and Functional Analyses of the Shedding Protease ADAM17 in HoxB8-Immortalized Macrophages and Dendritic-like Cells. J. Immunol. 2018, 201, 3106–3118. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.L.; French, K.S.; Resta, R.G.; Austin, J. Practice resource-focused revision: Standardized pedigree nomenclature update centered on sex and gender inclusivity: A practice resource of the National Society of Genetic Counselors. J. Genet. Couns. 2022, 31, 1238–1248. [Google Scholar] [CrossRef]
- Lin, P.-Y.; Chao, T.-C.; Wu, M.-L. Quantitative Susceptibility Mapping of Human Brain at 3T: A Multisite Reproducibility Study. Am. J. Neuroradiol. 2015, 36, 467–474. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Byrne, A.B.; Feng, B.J.; Pagel, K.A.; Mooney, S.D.; Karchin, R.; O’Donnell-Luria, A.; Harrison, S.M.; Tavtigian, S.V.; Greenblatt, M.S.; et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am. J. Hum. Genet. 2022, 109, 2163–2177. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Dick, K.J.; Eckhardt, M.; Paisán-Ruiz, C.; Alshehhi, A.A.; Proukakis, C.; Sibtain, N.A.; Maier, H.; Sharifi, R.; Patton, M.A.; Bashir, W.; et al. Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35). Hum. Mutat. 2010, 31, E1251–E1260. [Google Scholar] [CrossRef] [PubMed]
- Soehn, A.S.; Rattay, T.W.; Beck-Wödl, S.; Schäferhoff, K.; Monk, D.; Döbler-Neumann, M.; Hörtnagel, K.; Schlüter, A.; Ruiz, M.; Pujol, A.; et al. Uniparental disomy of chromosome 16 unmasks recessive mutations of FA2H/SPG35 in 4 families. Neurology 2016, 87, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Kruer, M.C.; Paisán-Ruiz, C.; Boddaert, N.; Yoon, M.Y.; Hama, H.; Gregory, A.; Malandrini, A.; Woltjer, R.L.; Munnich, A.; Gobin, S.; et al. Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann. Neurol. 2010, 68, 611–618. [Google Scholar] [CrossRef]
- Pensato, V.; Castellotti, B.; Gellera, C.; Pareyson, D.; Ciano, C.; Nanetti, L.; Salsano, E.; Piscosquito, G.; Sarto, E.; Eoli, M.; et al. Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain 2014, 137, 1907–1920. [Google Scholar] [CrossRef]
- Rupps, R.; Hukin, J.; Balicki, M.; Mercimek-Mahmutoglu, S.; Rolfs, A.; Dias, C. Novel Mutations in FA2H-Associated Neurodegeneration: An Underrecognized Condition? J. Child. Neurol. 2013, 28, 1500–1504. [Google Scholar] [CrossRef]
- Edvardson, S.; Hama, H.; Shaag, A.; Gomori, J.M.; Berger, I.; Soffer, D.; Korman, S.H.; Taustein, I.; Saada, A.; Elpeleg, O. Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia. Am. J. Hum. Genet. 2008, 83, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, N.; Abadi, R.N.S.; Alavi, A.; Rohani, M.; Ghasemi, A.; Tavasoli, A.R. The first reports of FA2H-associated neurodegeneration from two unrelated Iranian families. Neurol. Sci. 2023, 44, 4359–4362. [Google Scholar] [CrossRef] [PubMed]
- Incecik, F.; Besen, S.; Bozdogan, S.T. Hereditary Spastic Paraplegia Type 35 with a Novel Mutation in Fatty Acid 2-Hydroxylase Gene and Literature Review of the Clinical Features. Ann. Indian. Acad. Neurol. 2018, 21, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Bektaş, G.; Yeşil, G.; Yıldız, E.P.; Aydınlı, N.; Çalışkan, M.; Özmen, M. Hereditary spastic paraplegia type 35 caused by a novel FA2H mutation. Turk. J. Pediatr. 2017, 59, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Luo, Y.; Zhan, Z.; Du, J.; Hu, Z.; Wang, J.; Guo, J.; Hu, Z.; Yan, X.; Pan, Q.; et al. SPG35 contributes to the second common subtype of AR-HSP in China: Frequency analysis and functional characterization of FA2H gene mutations. Clin. Genet. 2015, 87, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.S.; Selim, L.; Mansour, L.; Mahmoud, I.G.; Fenstermaker, A.G.; Gabriel, S.B.; Gleeson, J.G. Mutations in FA2H in three Arab families with a clinical spectrum of neurodegeneration and hereditary spastic paraparesis. Clin. Genet. 2015, 88, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, A.; D’Angelo, M.G.; Arrigoni, F.; Brighina, E.; Arnoldi, A.; Citterio, A.; Bresolin, N.; Bassi, M.T. Atypical adult onset complicated spastic paraparesis with thin corpus callosum in two patients carrying a novel FA2H mutation. Eur. J. Neurol. 2012, 19, e127–e129. [Google Scholar] [CrossRef]
- Aguirre-Rodríguez, F.J.; Lucenilla, M.I.; Alvarez-Cubero, M.J.; Mata, C.; Entrala-Bernal, C.; Fernandez-Rosado, F. Novel FA2H mutation in a girl with familial spastic paraplegia. J. Neurol. Sci. 2015, 357, 332–334. [Google Scholar] [CrossRef]
- Pierson, T.M.; Simeonov, D.R.; Sincan, M.; Adams, D.A.; Markello, T.; Golas, G.; Fuentes-Fajardo, K.; Hansen, N.F.; Cherukuri, P.F.; Cruz, P.; et al. Exome sequencing and SNP analysis detect novel compound heterozygosity in fatty acid hydroxylase-associated neurodegeneration. Eur. J. Hum. Genet. 2012, 20, 476–479. [Google Scholar] [CrossRef]
- Cao, L.; Huang, X.J.; Chen, C.J.; Chen, S.D. A rare family with Hereditary Spastic Paraplegia Type 35 due to novel FA2H mutations: A case report with literature review. J. Neurol. Sci. 2013, 329, 1–5. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Article | FA2H Variant |
|---|---|
| [26] Dick | c.703C>T, c.159_176del18 |
| [2] Rattay | c.21delC, c.160_169del GCGGGCCAGG, c.205C>T, c.232G>A, c.262G>T, c.443C>T, c.503_506del TCTG, c.704G>A, c.859T>C, c.908G>T, c.956A>G |
| [27] Soehn | c.131C>A, c.133G>T, c.527G>A, c.785A>C |
| [28] Kruer | c.460C>T, c.510_511delCA |
| [29] Pensato | c.620C>T |
| [30] Rupps | c.209C>T, c.968C>T |
| [31] Edvardson | c.103G>T, c.786+1G>A |
| [10] Mari | c.193C>T, c.805c>T, c.1055C>T, c.1501A>G, [c.340_363del24][c.363+1_8del8] |
| [32] Hashemi | c.131delC |
| [33] Incecik | c.130C>T |
| [34] Bektaş | c.160_169dup |
| [35] Liao | c.388C>T, c.506+6C>G, c.230T>G |
| [36] Zaki | c.265C>T |
| [37] Tonelli | c.509A>G |
| [38] Aguirre-Rodriguez | C565C>T |
| [39] Pierson | c.707C>T |
| [40] Cao | c.968C>A; c.976G>A; c.688G>A |
| This report | c.75C>G |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
German, A.; Jukic, J.; Laner, A.; Arnold, P.; Socher, E.; Mennecke, A.; Schmidt, M.A.; Winkler, J.; Abicht, A.; Regensburger, M. Novel Homozygous FA2H Variant Causing the Full Spectrum of Fatty Acid Hydroxylase-Associated Neurodegeneration (SPG35). Genes 2024, 15, 14. https://doi.org/10.3390/genes15010014
German A, Jukic J, Laner A, Arnold P, Socher E, Mennecke A, Schmidt MA, Winkler J, Abicht A, Regensburger M. Novel Homozygous FA2H Variant Causing the Full Spectrum of Fatty Acid Hydroxylase-Associated Neurodegeneration (SPG35). Genes. 2024; 15(1):14. https://doi.org/10.3390/genes15010014
Chicago/Turabian StyleGerman, Alexander, Jelena Jukic, Andreas Laner, Philipp Arnold, Eileen Socher, Angelika Mennecke, Manuel A. Schmidt, Jürgen Winkler, Angela Abicht, and Martin Regensburger. 2024. "Novel Homozygous FA2H Variant Causing the Full Spectrum of Fatty Acid Hydroxylase-Associated Neurodegeneration (SPG35)" Genes 15, no. 1: 14. https://doi.org/10.3390/genes15010014
APA StyleGerman, A., Jukic, J., Laner, A., Arnold, P., Socher, E., Mennecke, A., Schmidt, M. A., Winkler, J., Abicht, A., & Regensburger, M. (2024). Novel Homozygous FA2H Variant Causing the Full Spectrum of Fatty Acid Hydroxylase-Associated Neurodegeneration (SPG35). Genes, 15(1), 14. https://doi.org/10.3390/genes15010014