
Differential Gene Expression Patterns in Peach Roots under Non-Uniform Soil Conditions in Response to Organic Matter


Abstract
:1. Introduction
2. Materials and Methods
2.1. Design and Treatments
2.2. RNA Extraction, Sequencing, and Data Analysis
2.3. Gene Ontology (GO)
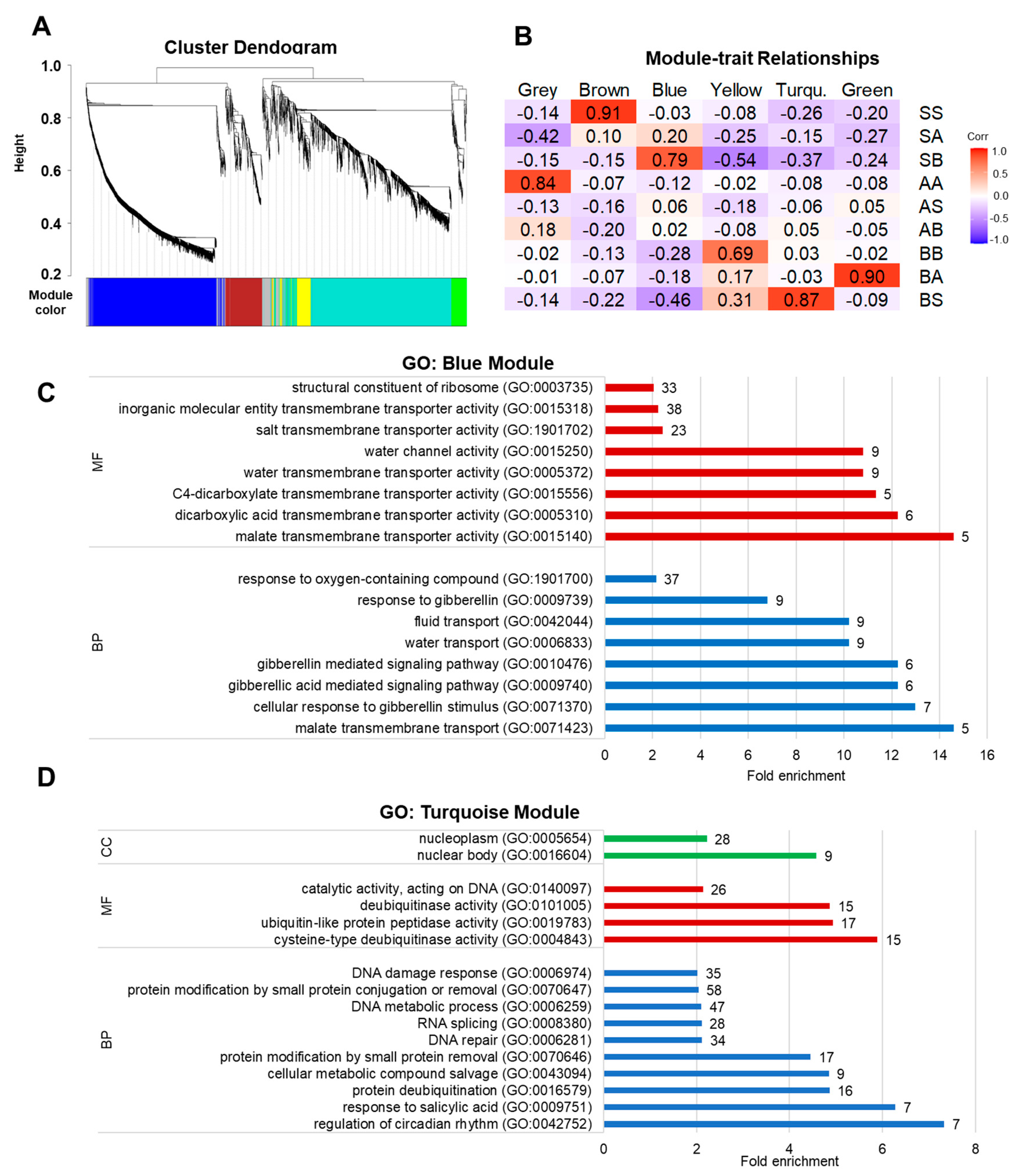
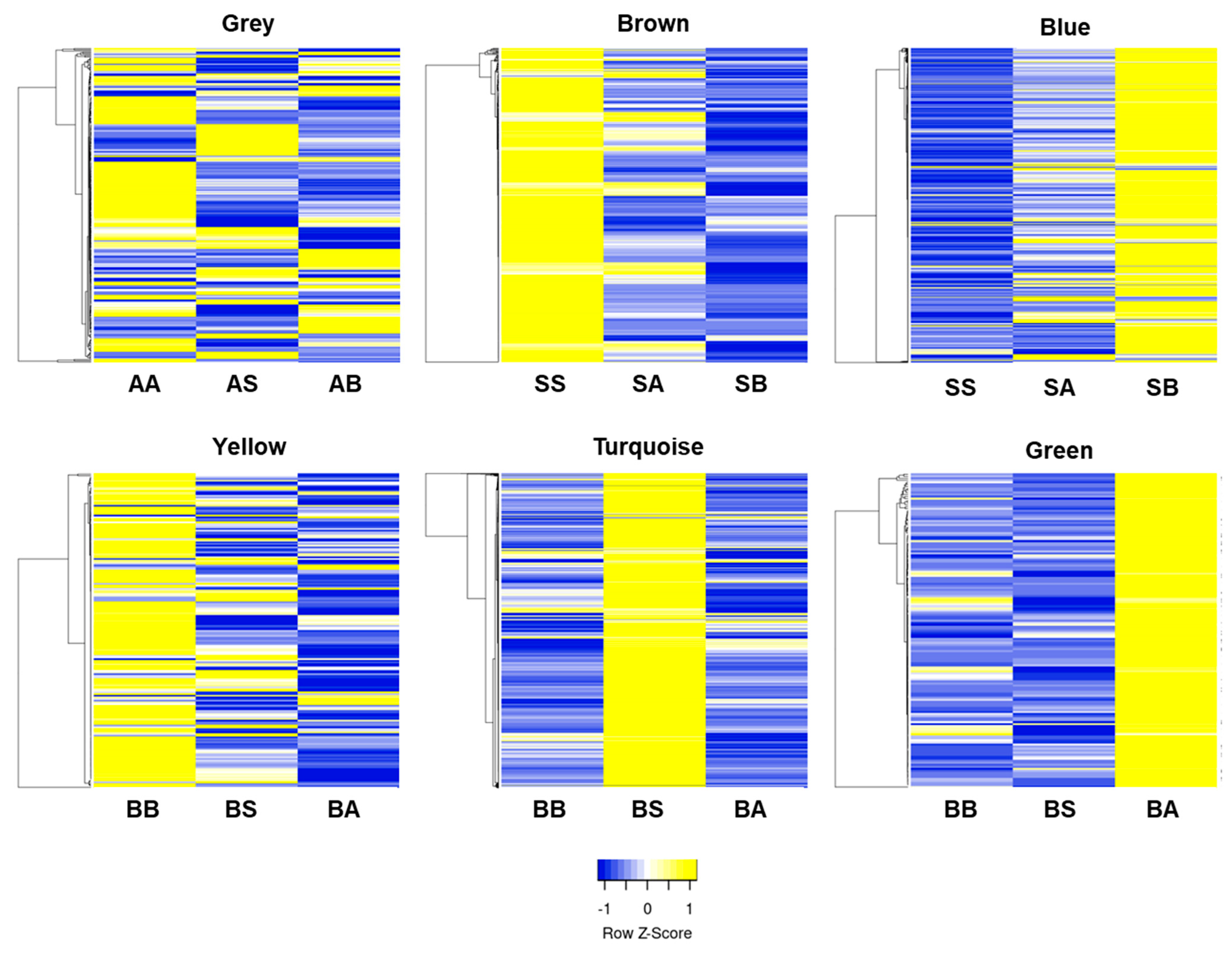
2.4. Co-Expression Network Analysis
2.5. Statistical Analysis
3. Results and Discussion
3.1. Plant Growth Prior to Root Sampling
3.2. Differential Gene Expression, Relative Expression, and Ontology
3.3. Clustering of DEGs by Modules
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UN. Transforming Our World: The 2030 Agenda for Sustainable Development; Resolution adopted by the General Assembly on 25 September 2015; United Nations: New York, NY, USA, 2015; 35p. [Google Scholar]
- Khalsa, S.D.S.; Hart, S.C.; Brown, P.H. Nutrient dynamics from surface-applied organic matter amendments on no-till orchard soil. Soil. Use Manag. 2022, 38, 649–662. [Google Scholar] [CrossRef]
- Chatzistathis, T.; Kavvadias, V.; Sotiropoulos, T.; Papadakis, I.E. Organic fertilization and tree orchards. Agriculture 2021, 11, 692. [Google Scholar] [CrossRef]
- Sofo, A.; Mininni, A.N.; Ricciuti, P. Soil macrofauna: A key factor for increasing soil fertility and promoting sustainable soil use in fruit orchard agrosystems. Agronomy 2020, 10, 456. [Google Scholar] [CrossRef]
- Sądej, W.; Żołnowski, A.C. Comparison of the effect of various long-term fertilization systems on the content and fractional composition of humic compounds in lessive soil. Plant Soil. Environ. 2019, 65, 172–180. [Google Scholar] [CrossRef]
- Liang, B.; Ma, C.; Fan, L.; Wang, Y.; Yuan, Y. Soil amendment alters soil physicochemical properties and bacterial community structure of a replanted apple orchard. Microbiol. Res. 2018, 216, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.M.; Zheng, F.L.; Abd_Allah, E.F.; Muthuramalingam, P.; Wu, Q.S.; Hashem, A. Spatial changes of arbuscular mycorrhizal fungi in peach and their correlation with soil properties. Saudi J. Biol. Sci. 2021, 28, 6495–6499. [Google Scholar] [CrossRef] [PubMed]
- Vives-Peris, V.; de Ollas, C.; Gómez-Cadenas, A.; Pérez-Clemente, R.M. Root exudates: From plant to rhizosphere and beyond. Plant Cell Rep. 2020, 39, 3–17. [Google Scholar] [CrossRef]
- Cervantes-Gámez, R.G.; Bueno-Ibarra, M.A.; Cruz-Mendívil, A.; Calderón-Vázquez, C.L.; Ramírez-Douriet, C.M.; Maldonado-Mendoza, I.E.; Villalobos-López, M.Á.; Valdez-Ortíz, Á.; López-Meyer, M. Arbuscular mycorrhizal symbiosis-induced expression changes in Solanum lycopersicum leaves revealed by RNA-seq analysis. Plant Mol. Biol. Rep. 2016, 34, 89–102. [Google Scholar] [CrossRef]
- Zouari, I.; Salvioli, A.; Chialva, M.; Novero, M.; Miozzi, L.; Tenore, G.C.; Bagnaresi, P.; Bonfante, P. From root to fruit: RNA-Seq analysis shows that arbuscular mycorrhizal symbiosis may affect tomato fruit metabolism. BMC Genom. 2014, 15, 1–19. [Google Scholar] [CrossRef]
- Doungous, O.; Minyaka, E.; Longue, E.A.M.; Nkengafac, N.J. Potentials of cocoa pod husk-based compost on Phytophthora pod rot disease suppression, soil fertility, and Theobroma cacao L. growth. Environ. Sci. Pollut. Res. 2018, 25, 25327–25335. [Google Scholar] [CrossRef]
- Ksouri, N.; Jiménez, S.; Wells, C.E.; Contreras-Moreira, B.; Gogorcena, Y. Transcriptional responses in root and leaf of Prunus persica under drought stress using RNA sequencing. Front. Plant Sci. 2016, 7, 1715. [Google Scholar] [CrossRef] [PubMed]
- González-Morales, S.; Solís-Gaona, S.; Valdés-Caballero, M.V.; Juárez-Maldonado, A.; Loredo-Treviño, A.; Benavides-Mendoza, A. Transcriptomics of biostimulation of plants under abiotic stress. Front. Genet. 2021, 12, 583888. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, J.; Zhou, X.; Zhao, J.; Liu, X.; Jiang, Q.; Ren, F. Transcriptome and metabolome studies on pre-harvest nitrogen impact on fruit yield and quality of peach (Prunus persica L.). Metabolites 2022, 12, 905. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Zhang, J.; Zhang, Q.; Li, X.; Li, M.; Yang, Y.; Zhou, J.; Wei, Q.; Zhou, B. Transcriptome and metabolome analyses revealed the response mechanism of apple to different phosphorus stresses. Plant Physiol. Biochem. 2021, 167, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Montanaro, G.; Xiloyannis, C.; Nuzzo, V.; Dichio, B. Orchard management, soil organic carbon and ecosystem services in Mediterranean fruit tree crops. Sci. Hortic. 2017, 217, 92–101. [Google Scholar] [CrossRef]
- Reganold, J.P.; Glover, J.D.; Andrews, P.K.; Hinman, H.R. Sustainability of three apple production systems. Nature 2001, 410, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Arús, P.; Verde, I.; Sosinski, B.; Zhebentyayeva, T.; Abbott, A.G. The peach genome. Tree Genet. Genomes 2012, 8, 531–547. [Google Scholar] [CrossRef]
- Lawson, J.D.; Bridges, W.C.; Adelberg, J.W. IBA delivery technique and media salts affected in vitro rooting and acclimatization of eight Prunus genotypes. Plants 2023, 12, 289. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinform. 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Jung, S.; Lee, T.; Cheng, C.H.; Buble, K.; Zheng, P.; Yu, J.; Humann, J.; Ficklin, S.P.; Gasic, K.; Scott, K.; et al. 15 years of GDR: New data and functionality in the Genome Database for Rosaceae. Nucleic Acids Res. 2019, 47, D1137–D1145. [Google Scholar] [CrossRef]
- Verde, I.; Abbott, A.G.; Scalabrin, S.; Jung, S.; Shu, S.; Marroni, F.; Zhebentyayeva, T.; Dettori, M.T.; Grimwood, J.; Cattonaro, F.; et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013, 45, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Smid, M.; Coebergh van den Braak, R.R.; van de Werken, H.J.; van Riet, J.; van Galen, A.; de Weerd, V.; van der Vlugt-Daane, M.; Bril, S.I.; Lalmahomed, Z.S.; Kloosterman, W.P.; et al. Gene length corrected trimmed mean of M-values (GeTMM) processing of RNA-seq data performs similarly in intersample analyses while improving intrasample comparisons. BMC Bioinform. 2018, 19, 236. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]
- Gene Ontology Database. Available online: http://www.geneontology.org (accessed on 15 June 2023).
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Baldi, E.; Toselli, M.; Eissenstat, D.M.; Marangoni, B. Organic fertilization leads to increased peach root production and lifespan. Tree Physiol. 2010, 30, 1373–1382. [Google Scholar] [CrossRef]
- Chauhan, A.; Saini, R.; Sharma, J.C. Plant growth promoting rhizobacteria and their biological properties for soil enrichment and growth promotion. J. Plant Nutr. 2021, 45, 273–299. [Google Scholar] [CrossRef]
- Cavender, N.D.; Atiyeh, R.M.; Knee, M. Vermicompost stimulates mycorrhizal colonization of roots of Sorghum bicolor at the expense of plant growth. Pedobiologia 2003, 47, 85–89. [Google Scholar] [CrossRef]
- Schmitz, A.M.; Harrison, M.J. Signaling events during initiation of arbuscular mycorrhizal symbiosis. J. Integr. Plant Biol. 2014, 56, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Ali, S.; Zhang, H.; Ouyang, Y.; Qiu, B.; Wu, F.; Zhang, G. The influence of pH and organic matter content in paddy soil on heavy metal availability and their uptake by rice plants. Environ. Pollut. 2011, 159, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.; Singh, P.K.; Mankotia, S.; Swain, J.; Satbhai, S.B. Iron homeostasis in plants and its crosstalk with copper, zinc, and manganese. Plant Stress. 2021, 1, 100008. [Google Scholar] [CrossRef]
- Skuza, L.; Szućko-Kociuba, I.; Filip, E.; Bożek, I. Natural molecular mechanisms of plant hyperaccumulation and hypertolerance towards heavy metals. Int. J. Mol. Sci. 2022, 23, 9335. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Barragan, B.A.; Kusakabe, A.; Melgar, J.C.; Nelson, S.D. Understanding partial rootzone drying in citrus. Acta Hortic. 2016, 1135, 123–130. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, Y.; Zhong, J.; Zhang, T.; Li, D.; Ba, T.; Xu, T.; Chang, L.; Zhang, Q.; Sun, M. Root physiological traits and transcriptome analyses reveal that root zone water retention confers drought tolerance to Opisthopappus taihangensis. Sci. Rep. 2020, 10, 2627. [Google Scholar] [CrossRef]
- Chen, H.; Yang, R.; Zhang, X.; Chen, Y.; Xia, Y.; Xu, X. Foliar application of gibberellin inhibits the cadmium uptake and xylem transport in lettuce (Lactuca sativa L.). Sci. Hortic. 2021, 288, 110410. [Google Scholar] [CrossRef]
- French, E.; Iyer-Pascuzzi, A.S. A role for the gibberellin pathway in biochar-mediated growth promotion. Sci. Rep. 2018, 8, 5389. [Google Scholar] [CrossRef]
- Veach, A.M.; Morris, R.; Yip, D.Z.; Yang, Z.K.; Engle, N.L.; Cregger, M.A.; Tschaplinski, T.J.; Schadt, C.W. Rhizosphere microbiomes diverge among Populus trichocarpa plant-host genotypes and chemotypes, but it depends on soil origin. Microbiome 2019, 7, 76. [Google Scholar] [CrossRef]
- Hubbard, C.J.; McMinn, R.; Weinig, C. Rhizosphere microbes influence host circadian clock function. Phytobiomes J. 2021, 5, 368–372. [Google Scholar] [CrossRef]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Nutrient or Characteristic | Value |
|---|---|
| Total N (g kg−1) | 8.3 |
| P (g kg−1) | 2.0 |
| K (g kg−1) | 2.2 |
| Ca (g kg−1) | 8.5 |
| Mg (g kg−1) | 1.4 |
| S (g kg−1) | 0.08 |
| Zn (mg kg−1) | 0.04 |
| Cu (mg kg−1) | 0.02 |
| Mn (mg kg−1) | 0.16 |
| Fe (mg kg−1) | 5.14 |
| Na (mg kg−1) | 0.71 |
| Al (mg kg−1) | 5.70 |
| C:N (ratio) | 11.7 |
| OM (%) | 19.4 |
| EC (dS m−1) | 0.63 |
| pH | 7.50 |
| Treatment Code | Soil Where Root Tissue Was Sampled | Adjacent Soil | Adjacent Soil Factor |
|---|---|---|---|
| SS | S | S | n/a |
| AA | A | A | n/a |
| BB | B | B | n/a |
| SA | S | A | + OM |
| SB | S | B | + soil life and OM |
| AS | A | S | − OM |
| AB | A | B | + soil life |
| BS | B | S | − soil life and OM |
| BA | B | A | − soil life |
| Treatment | Length | Area | Avg Dia | Root Volume | Tips | Forks | Crossings |
|---|---|---|---|---|---|---|---|
| AA | 1845.9 | 458.1 | 0.8 | 9.0 | 3569 | 6602 | 1266 |
| BB | 711.5 | 556.1 | 2.5 | 34.6 | 1594 | 3219 | 667 |
| SS | 1658.4 | 358.3 | 0.7 | 6.2 | 2978 | 7882 | 2259 |
| Group | Comparison | Adjacent Factor | Total | Down | Up |
|---|---|---|---|---|---|
| SS vs. AA | OM | 317 | 188 | 129 | |
| Uniform | AA vs. BB | soil life | 121 | 53 | 68 |
| SS vs. BB | OM and soil life | 370 | 176 | 194 | |
| S | SA vs. SS | OM | 200 | 98 | 102 |
| − soil life | SA vs. SB | soil life | 221 | 63 | 158 |
| − OM | SB vs. SS | OM and soil life | 411 | 274 | 137 |
| A | AS vs. AA | OM | 94 | 52 | 42 |
| − soil life | AB vs. AA | soil life | 83 | 53 | 30 |
| + OM | AS vs. AB | OM and soil life | 19 | 8 | 11 |
| B | BS vs. BA | OM | 332 | 193 | 139 |
| + soil life | BA vs. BB | soil life | 106 | 33 | 73 |
| + OM | BS vs. BB | OM and soil life | 239 | 54 | 185 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lawrence, B.T.; Calle, A.; Saski, C.A.; Melgar, J.C. Differential Gene Expression Patterns in Peach Roots under Non-Uniform Soil Conditions in Response to Organic Matter. Genes 2024, 15, 70. https://doi.org/10.3390/genes15010070
Lawrence BT, Calle A, Saski CA, Melgar JC. Differential Gene Expression Patterns in Peach Roots under Non-Uniform Soil Conditions in Response to Organic Matter. Genes. 2024; 15(1):70. https://doi.org/10.3390/genes15010070
Chicago/Turabian StyleLawrence, Brian T., Alejandro Calle, Christopher A. Saski, and Juan Carlos Melgar. 2024. "Differential Gene Expression Patterns in Peach Roots under Non-Uniform Soil Conditions in Response to Organic Matter" Genes 15, no. 1: 70. https://doi.org/10.3390/genes15010070