
Dynamic Evolution of Repetitive Elements and Chromatin States in Apis mellifera Subspecies

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. RE Identification
2.2. RNA-seq Analysis
2.3. Genome Segmentation with ChromHMM
3. Results
3.1. Expansion of Previously Uncharacterized Repeat Families Discriminate the A. m. mellifera and A. m. ligustica Subspecies
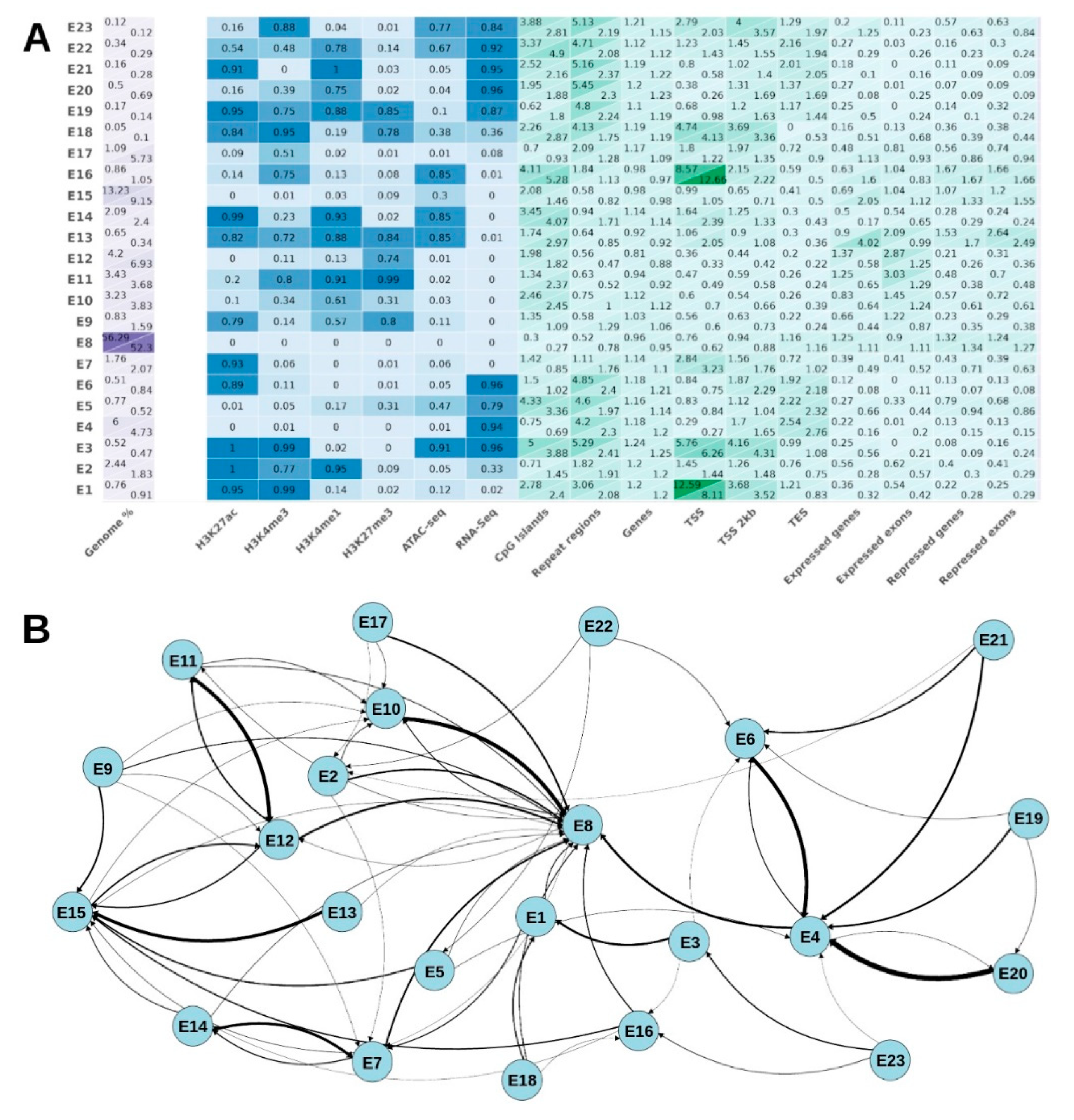
3.2. Integrating Multi-Omics Data via Markov Models for Genome-Wide Chromatin State Assignment
3.3. REs Are Significantly Enriched in Special Chromatin State (E13)
3.4. REs Demonstrate Opposing Trends in E8 and E15 Chromatin State Proportions within 0–3 and 20–40 Kimura Distances
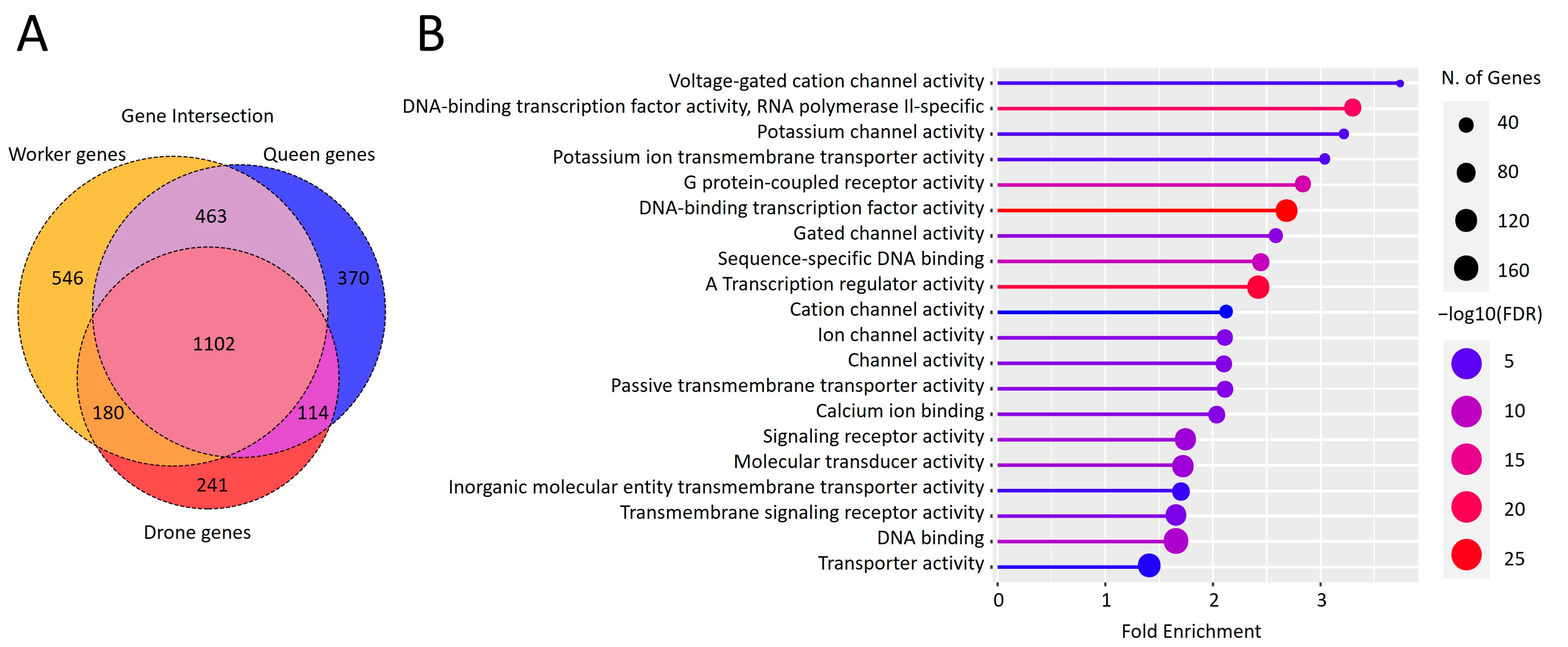
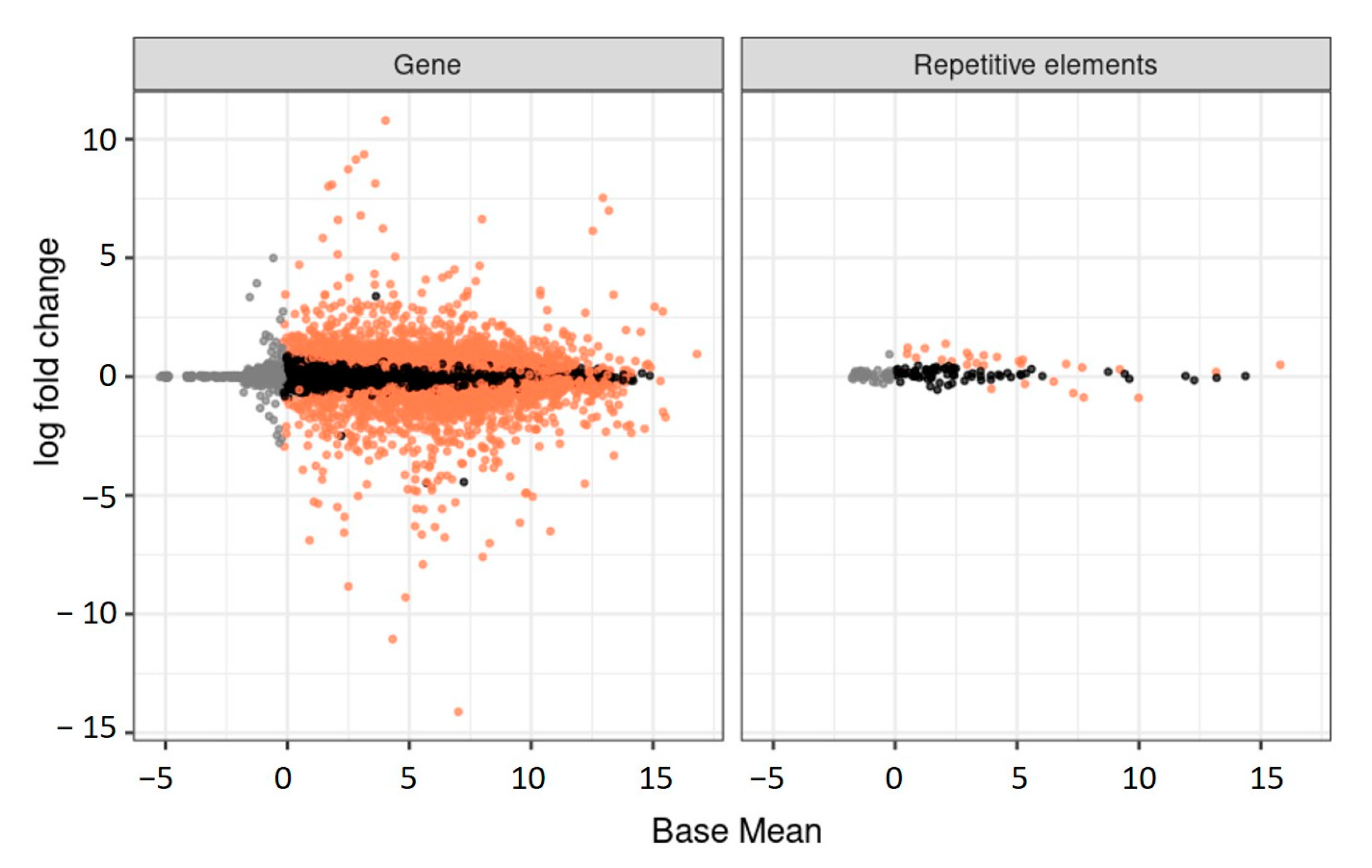
3.5. Transcriptional Activity of Repetitive Elements during the Larval Stage in Queens and Workers
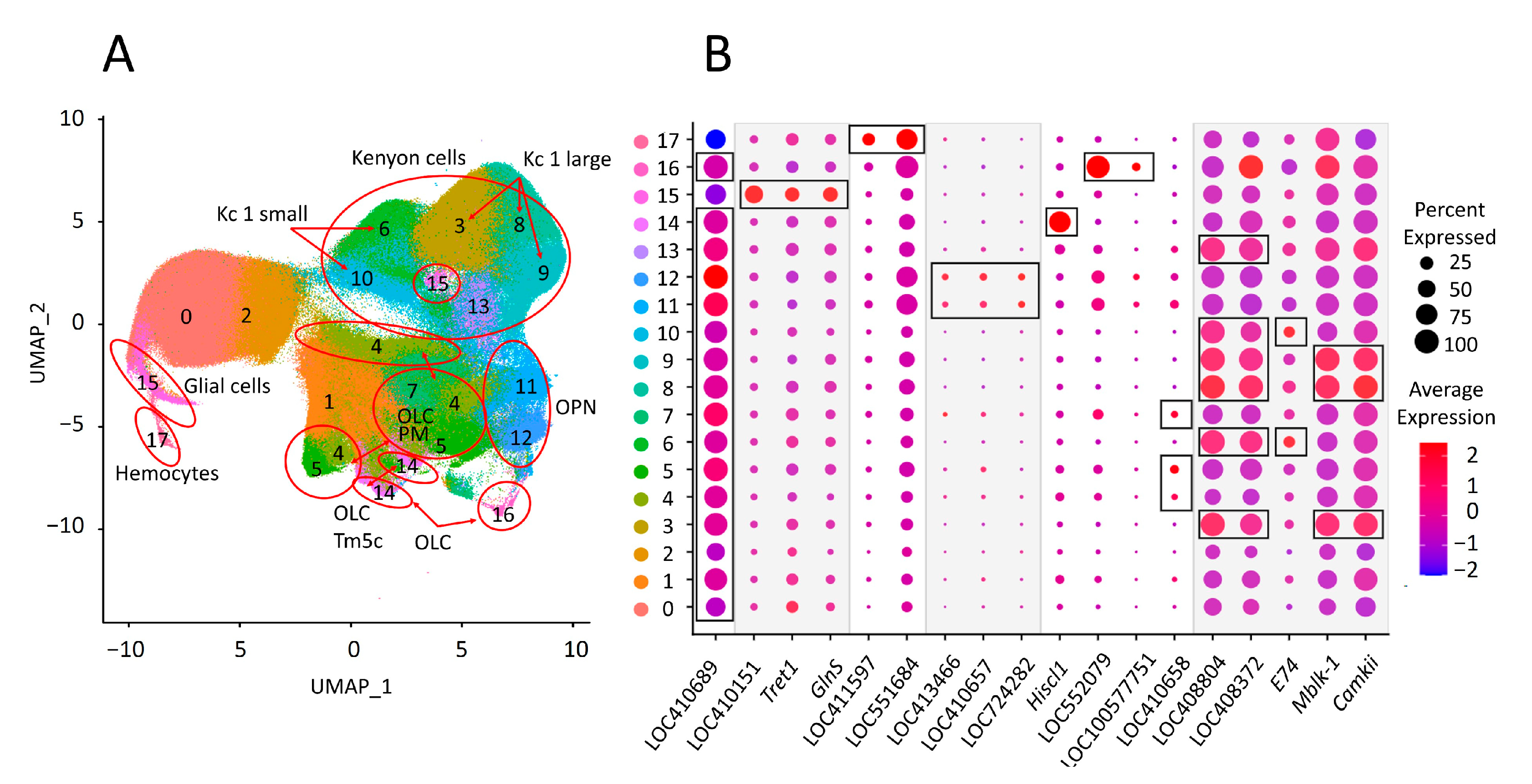
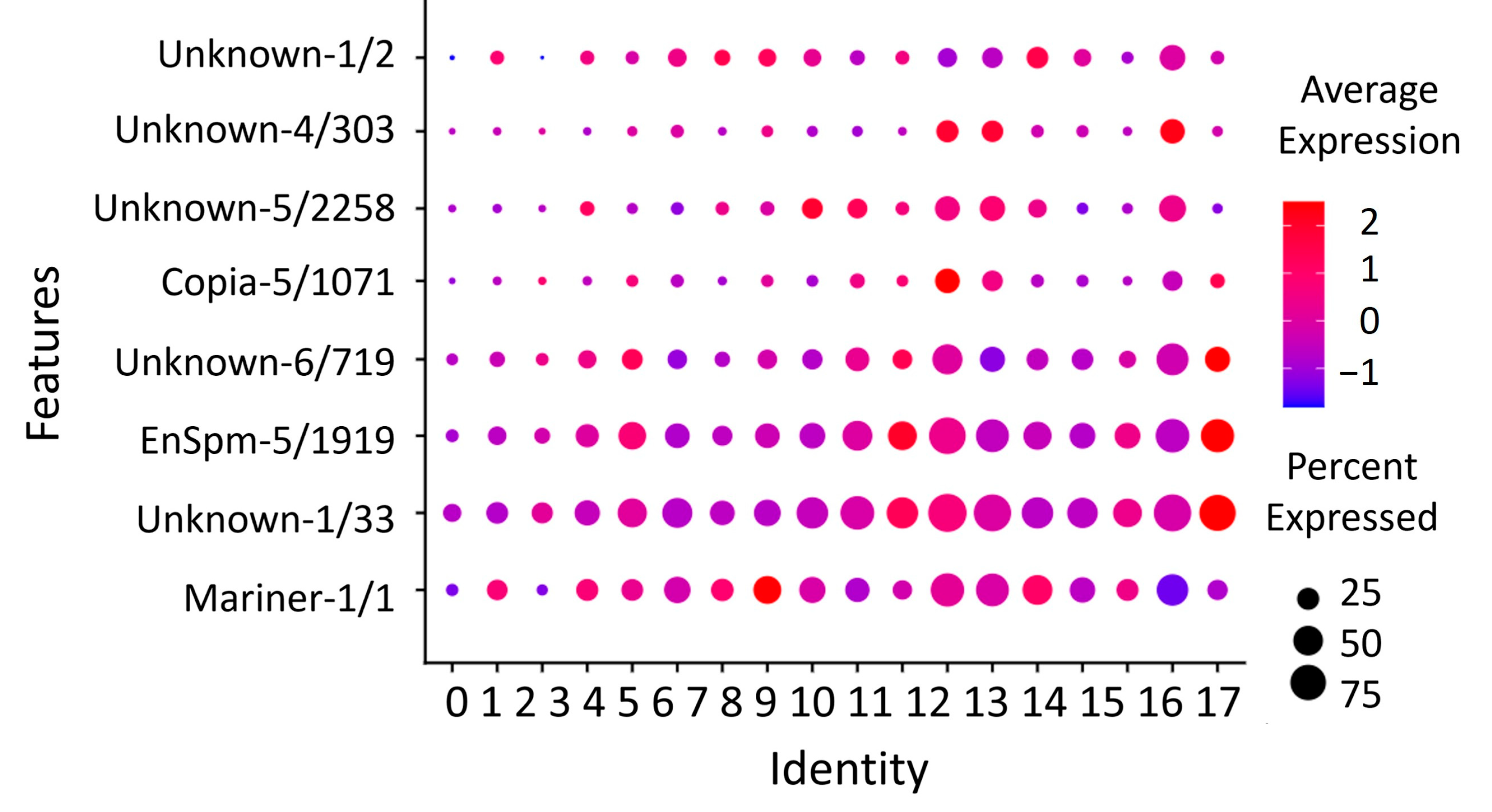
3.6. Expression of Eight Repeats Segregates Brain Cell Populations
3.7. Repetitive Elements
4. Discussion
4.1. Eusociality
4.2. Distribution of TEs
4.3. Limitations of ChromHMM
4.4. REs Markers in scRNA-seq Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aizen, M.A.; Garibaldi, L.A.; Cunningham, S.A.; Klein, A.M. How Much Does Agriculture Depend on Pollinators? Lessons from Long-Term Trends in Crop Production. Ann. Bot. 2009, 103, 1579–1588. [Google Scholar] [CrossRef]
- Christmann, S. Do We Realize the Full Impact of Pollinator Loss on Other Ecosystem Services and the Challenges for Any Restoration in Terrestrial Areas? Restor. Ecol. 2019, 27, 720–725. [Google Scholar] [CrossRef]
- Patel, V.; Pauli, N.; Biggs, E.; Barbour, L.; Boruff, B. Why Bees Are Critical for Achieving Sustainable Development. Ambio 2021, 50, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Dangles, O.; Casas, J. Ecosystem Services Provided by Insects for Achieving Sustainable Development Goals. Ecosyst. Serv. 2019, 35, 109–115. [Google Scholar] [CrossRef]
- Kohno, H.; Kubo, T. Genetics in the Honey Bee: Achievements and Prospects toward the Functional Analysis of Molecular and Neural Mechanisms Underlying Social Behaviors. Insects 2019, 10, 348. [Google Scholar] [CrossRef]
- Wilson, E.O.; Hölldobler, B. Eusociality: Origin and Consequences. Proc. Natl. Acad. Sci. USA 2005, 102, 13367–13371. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J. Life History and the Transitions to Eusociality in the Hymenoptera. Front. Ecol. Evol. 2021, 9. [Google Scholar] [CrossRef]
- Ashby, R.; Forêt, S.; Searle, I.; Maleszka, R. MicroRNAs in Honey Bee Caste Determination. Sci. Rep. 2016, 6, 18794. [Google Scholar] [CrossRef]
- Alaux, C.; Sinha, S.; Hasadsri, L.; Hunt, G.J.; Guzmán-Novoa, E.; DeGrandi-Hoffman, G.; Uribe-Rubio, J.L.; Southey, B.R.; Rodriguez-Zas, S.; Robinson, G.E. Honey Bee Aggression Supports a Link between Gene Regulation and Behavioral Evolution. Proc. Natl. Acad. Sci. USA 2009, 106, 15400–15405. [Google Scholar] [CrossRef]
- Greenberg, J.K.; Xia, J.; Zhou, X.; Thatcher, S.R.; Gu, X.; Ament, S.A.; Newman, T.C.; Green, P.J.; Zhang, W.; Robinson, G.E.; et al. Behavioral Plasticity in Honey Bees Is Associated with Differences in Brain microRNA Transcriptome. Genes Brain Behav. 2012, 11, 660–670. [Google Scholar] [CrossRef]
- Eyer, M.; Dainat, B.; Neumann, P.; Dietemann, V. Social Regulation of Ageing by Young Workers in the Honey Bee, Apis mellifera. Exp. Gerontol. 2017, 87, 84–91. [Google Scholar] [CrossRef] [PubMed]
- de Paula Junior, D.E.; de Oliveira, M.T.; Bruscadin, J.J.; Pinheiro, D.G.; Bomtorin, A.D.; Coelho Júnior, V.G.; Moda, L.M.R.; Simões, Z.L.P.; Barchuk, A.R. Caste-Specific Gene Expression Underlying the Differential Adult Brain Development in the Honeybee Apis mellifera. Insect Mol. Biol. 2021, 30, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xiao, Y.; Li, Y.; Wang, X.; Qi, S.; Wang, Y.; Zhao, L.; Wang, K.; Peng, W.; Luo, G.-Z.; et al. RNA m6A Modification Functions in Larval Development and Caste Differentiation in Honeybee (Apis mellifera). Cell Rep. 2021, 34, 108580. [Google Scholar] [CrossRef]
- Yokoi, K.; Wakamiya, T.; Bono, H. Meta-Analysis of the Public RNA-Seq Data of the Western Honeybee Apis mellifera to Construct Reference Transcriptome Data. Insects 2022, 13, 931. [Google Scholar] [CrossRef] [PubMed]
- Gregory, T.R.; Nicol, J.A.; Tamm, H.; Kullman, B.; Kullman, K.; Leitch, I.J.; Murray, B.G.; Kapraun, D.F.; Greilhuber, J.; Bennett, M.D. Eukaryotic Genome Size Databases. Nucleic Acids Res. 2007, 35, D332–D338. [Google Scholar] [CrossRef]
- Petersen, M.; Armisén, D.; Gibbs, R.A.; Hering, L.; Khila, A.; Mayer, G.; Richards, S.; Niehuis, O.; Misof, B. Diversity and Evolution of the Transposable Element Repertoire in Arthropods with Particular Reference to Insects. BMC Evol. Biol. 2019, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Yang, M.; Guo, W.; Wang, X.; Kang, L. Large-Scale Transcriptome Analysis of Retroelements in the Migratory Locust, Locusta Migratoria. PLoS ONE 2012, 7, e40532. [Google Scholar] [CrossRef]
- Gilbert, C.; Peccoud, J.; Cordaux, R. Transposable Elements and the Evolution of Insects. Annu. Rev. Entomol. 2021, 66, 355–372. [Google Scholar] [CrossRef]
- Feschotte, C. Transposable Elements and the Evolution of Regulatory Networks. Nat. Rev. Genet. 2008, 9, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten Things You Should Know about Transposable Elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef]
- Carareto, C.M.A.; Hernandez, E.H.; Vieira, C. Genomic Regions Harboring Insecticide Resistance-Associated Cyp Genes Are Enriched by Transposable Element Fragments Carrying Putative Transcription Factor Binding Sites in Two Sibling Drosophila Species. Gene 2014, 537, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Lu, J. Diversification of Transposable Elements in Arthropods and Its Impact on Genome Evolution. Genes 2019, 10, 338. [Google Scholar] [CrossRef] [PubMed]
- Ellison, C.E.; Bachtrog, D. Dosage Compensation via Transposable Element Mediated Rewiring of a Regulatory Network. Science 2013, 342, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Pardue, M.-L.; Rashkova, S.; Casacuberta, E.; DeBaryshe, P.G.; George, J.A.; Traverse, K.L. Two Retrotransposons Maintain Telomeres in Drosophila. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2005, 13, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Jangam, D.; Feschotte, C.; Betrán, E. Transposable Element Domestication as an Adaptation to Evolutionary Conflicts. Trends Genet. TIG 2017, 33, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Guigó, R. The RIDL Hypothesis: Transposable Elements as Functional Domains of Long Noncoding RNAs. RNA 2014, 20, 959–976. [Google Scholar] [CrossRef] [PubMed]
- Bonasio, R. Emerging Topics in Epigenetics: Ants, Brains, and Noncoding RNAs. Ann. N. Y. Acad. Sci. 2012, 1260, 14–23. [Google Scholar] [CrossRef]
- Bonasio, R.; Shiekhattar, R. Regulation of Transcription by Long Noncoding RNAs. Annu. Rev. Genet. 2014, 48, 433–455. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome Regulation by Long Noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef]
- Hou, Y.; Zhang, R.; Sun, X. Enhancer LncRNAs Influence Chromatin Interactions in Different Ways. Front. Genet. 2019, 10, 936. [Google Scholar] [CrossRef] [PubMed]
- Amaral, P.P.; Leonardi, T.; Han, N.; Viré, E.; Gascoigne, D.K.; Arias-Carrasco, R.; Büscher, M.; Pandolfini, L.; Zhang, A.; Pluchino, S.; et al. Genomic Positional Conservation Identifies Topological Anchor Point RNAs Linked to Developmental Loci. Genome Biol. 2018, 19, 32. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Haines, J.E.; Perez, E.M.; Munson, G.; Chen, J.; Kane, M.; McDonel, P.E.; Guttman, M.; Lander, E.S. Local Regulation of Gene Expression by lncRNA Promoters, Transcription and Splicing. Nature 2016, 539, 452–455. [Google Scholar] [CrossRef]
- Feng, W.; Huang, J.; Zhang, Z.; Nie, H.; Lin, Y.; Li, Z.; Su, S. Understanding of Waggle Dance in the Honey Bee (Apis mellifera) from the Perspective of Long Non-Coding RNA. Insects 2022, 13, 111. [Google Scholar] [CrossRef]
- Liu, F.; Shi, T.; Qi, L.; Su, X.; Wang, D.; Dong, J.; Huang, Z.Y. lncRNA Profile of Apis mellifera and Its Possible Role in Behavioural Transition from Nurses to Foragers. BMC Genom. 2019, 20, 393. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.E.R.; Jones, B.M.; Hunt, B.G.; Kocher, S.D. Rate Variation in the Evolution of Non-Coding DNA Associated with Social Evolution in Bees. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180247. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowski, M.; Lowe, R.; Maleszka, J.; Conn, D.; Maleszka, R.; Hurd, P.J. Phenotypically Distinct Female Castes in Honey Bees Are Defined by Alternative Chromatin States during Larval Development. Genome Res. 2018, 28, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, L.; Zhao, Y.; Wang, Y.; Chen, C.; Hu, Y.; Zhu, Y.; Sun, H.; Cheng, Y.; Sun, Q.; et al. Single-Cell Transcriptomic Analysis of Honeybee Brains Identifies Vitellogenin as Caste Differentiation-Related Factor. iScience 2022, 25, 104643. [Google Scholar] [CrossRef] [PubMed]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial Reconstruction of Single-Cell Gene Expression Data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Lowe, R.; Wojciechowski, M.; Ellis, N.; Hurd, P.J. Chromatin Accessibility-Based Characterisation of Brain Gene Regulatory Networks in Three Distinct Honey Bee Polyphenisms. Nucleic Acids Res. 2022, 50, 11550–11562. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kellis, M. Chromatin-State Discovery and Genome Annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef] [PubMed]
- Kharchenko, P.V.; Alekseyenko, A.A.; Schwartz, Y.B.; Minoda, A.; Riddle, N.C.; Ernst, J.; Sabo, P.J.; Larschan, E.; Gorchakov, A.A.; Gu, T.; et al. Comprehensive Analysis of the Chromatin Landscape in Drosophila Melanogaster. Nature 2011, 471, 480–485. [Google Scholar] [CrossRef]
- Rada-Iglesias, A. Is H3K4me1 at Enhancers Correlative or Causative? Nat. Genet. 2018, 50, 4–5. [Google Scholar] [CrossRef]
- Barral, A.; Déjardin, J. The Chromatin Signatures of Enhancers and Their Dynamic Regulation. Nucleus 2023, 14, 2160551. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.N.; Meinertzhagen, I.A. The Functional Organisation of Glia in the Adult Brain of Drosophila and Other Insects. Prog. Neurobiol. 2010, 90, 471–497. [Google Scholar] [CrossRef]
- Allen, A.M.; Neville, M.C.; Birtles, S.; Croset, V.; Treiber, C.D.; Waddell, S.; Goodwin, S.F. A Single-Cell Transcriptomic Atlas of the Adult Drosophila Ventral Nerve Cord. eLife 2020, 9, e54074. [Google Scholar] [CrossRef]
- Davie, K.; Janssens, J.; Koldere, D.; De Waegeneer, M.; Pech, U.; Kreft, Ł.; Aibar, S.; Makhzami, S.; Christiaens, V.; Bravo González-Blas, C.; et al. A Single-Cell Transcriptome Atlas of the Aging Drosophila Brain. Cell 2018, 174, 982–998.e20. [Google Scholar] [CrossRef]
- Robinow, S.; White, K. Characterization and Spatial Distribution of the ELAV Protein during Drosophila Melanogaster Development. J. Neurobiol. 1991, 22, 443–461. [Google Scholar] [CrossRef]
- Galizia, C.G.; Menzel, R. Odour Perception in Honeybees: Coding Information in Glomerular Patterns. Curr. Opin. Neurobiol. 2000, 10, 504–510. [Google Scholar] [CrossRef]
- Groh, C.; Rössler, W. Analysis of Synaptic Microcircuits in the Mushroom Bodies of the Honeybee. Insects 2020, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Roat, T.C.; da Cruz-Landim, C. Mitosis and Cell Death in the Optic Lobes of Workers, Queens and Drones of the Honey Bee (Apis mellifera) during Metamorphosis. J. Biosci. 2010, 35, 415–425. [Google Scholar] [CrossRef]
- Caron, S.; Abbott, L.F. Neuroscience: Intelligence in the Honeybee Mushroom Body. Curr. Biol. CB 2017, 27, R220–R223. [Google Scholar] [CrossRef] [PubMed]
- Suenami, S.; Oya, S.; Kohno, H.; Kubo, T. Kenyon Cell Subtypes/Populations in the Honeybee Mushroom Bodies: Possible Function Based on Their Gene Expression Profiles, Differentiation, Possible Evolution, and Application of Genome Editing. Front. Psychol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Suenami, S.; Kubo, T. Gene Expression Profiles and Neural Activities of Kenyon Cell Subtypes in the Honeybee Brain: Identification of Novel “middle-Type” Kenyon Cells. Zool. Lett. 2016, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Eleftherianos, I.; Xu, M.; Yadi, H.; Ffrench-Constant, R.H.; Reynolds, S.E. Plasmatocyte-Spreading Peptide (PSP) Plays a Central Role in Insect Cellular Immune Defenses against Bacterial Infection. J. Exp. Biol. 2009, 212, 1840–1848. [Google Scholar] [CrossRef]
- Negri, P.; Maggi, M.; Ramirez, L.; Szawarski, N.; De Feudis, L.; Lamattina, L.; Eguaras, M. Cellular Immunity in Apis mellifera: Studying Hemocytes Brings Light about Bees Skills to Confront Threats. Apidologie 2016, 47, 379–388. [Google Scholar] [CrossRef]
- Chak, S.T.C.; Harris, S.E.; Hultgren, K.M.; Jeffery, N.W.; Rubenstein, D.R. Eusociality in Snapping Shrimps Is Associated with Larger Genomes and an Accumulation of Transposable Elements. Proc. Natl. Acad. Sci. USA 2021, 118, e2025051118. [Google Scholar] [CrossRef]
- Thorne, B.L.; Breisch, N.L.; Muscedere, M.L. Evolution of Eusociality and the Soldier Caste in Termites: Influence of Intraspecific Competition and Accelerated Inheritance. Proc. Natl. Acad. Sci. USA 2003, 100, 12808–12813. [Google Scholar] [CrossRef]
- Lawson, S.P.; Legan, A.W.; Graham, C.; Abbot, P. Comparative Phenotyping across a Social Transition in Aphids. Anim. Behav. 2014, 96, 117–125. [Google Scholar] [CrossRef]
- Chapman, T.W.; Crespi, B.J.; Kranz, B.D.; Schwarz, M.P. High Relatedness and Inbreeding at the Origin of Eusociality in Gall-Inducing Thrips. Proc. Natl. Acad. Sci. USA 2000, 97, 1648–1650. [Google Scholar] [CrossRef]
- Biedermann, P.H.W.; Taborsky, M. Larval Helpers and Age Polyethism in Ambrosia Beetles. Proc. Natl. Acad. Sci. USA 2011, 108, 17064–17069. [Google Scholar] [CrossRef] [PubMed]
- Oeyen, J.P.; Baa-Puyoulet, P.; Benoit, J.B.; Beukeboom, L.W.; Bornberg-Bauer, E.; Buttstedt, A.; Calevro, F.; Cash, E.I.; Chao, H.; Charles, H.; et al. Sawfly Genomes Reveal Evolutionary Acquisitions That Fostered the Mega-Radiation of Parasitoid and Eusocial Hymenoptera. Genome Biol. Evol. 2020, 12, 1099–1188. [Google Scholar] [CrossRef]
- Torres, V.O.; Montagna, T.S.; Raizer, J.; Antonialli-Junior, W.F. Division of Labor in Colonies of the Eusocial Wasp, Mischocyttarus Consimilis. J. Insect Sci. 2012, 12, 21. [Google Scholar] [CrossRef]
- Cardinal, S.; Danforth, B.N. The Antiquity and Evolutionary History of Social Behavior in Bees. PLoS ONE 2011, 6, e21086. [Google Scholar] [CrossRef]
- Favreau, E.; Martínez-Ruiz, C.; Rodrigues Santiago, L.; Hammond, R.L.; Wurm, Y. Genes and Genomic Processes Underpinning the Social Lives of Ants. Curr. Opin. Insect Sci. 2018, 25, 83–90. [Google Scholar] [CrossRef]
- Opachaloemphan, C.; Yan, H.; Leibholz, A.; Desplan, C.; Reinberg, D. Recent Advances in Behavioral (Epi)Genetics in Eusocial Insects. Annu. Rev. Genet. 2018, 52, 489–510. [Google Scholar] [CrossRef]
- Berger, J.; Legendre, F.; Zelosko, K.-M.; Harrison, M.C.; Grandcolas, P.; Bornberg-Bauer, E.; Fouks, B. Eusocial Transition in Blattodea: Transposable Elements and Shifts of Gene Expression. Genes 2022, 13, 1948. [Google Scholar] [CrossRef]
- Korb, J.; Poulsen, M.; Hu, H.; Li, C.; Boomsma, J.J.; Zhang, G.; Liebig, J. A Genomic Comparison of Two Termites with Different Social Complexity. Front. Genet. 2015, 6, 9. [Google Scholar] [CrossRef]
- Kapheim, K.M.; Pan, H.; Li, C.; Salzberg, S.L.; Puiu, D.; Magoc, T.; Robertson, H.M.; Hudson, M.E.; Venkat, A.; Fischman, B.J.; et al. Genomic Signatures of Evolutionary Transitions from Solitary to Group Living. Science 2015, 348, 1139–1143. [Google Scholar] [CrossRef]
- Fouks, B.; Brand, P.; Nguyen, H.N.; Herman, J.; Camara, F.; Ence, D.; Hagen, D.E.; Hoff, K.J.; Nachweide, S.; Romoth, L.; et al. The Genomic Basis of Evolutionary Differentiation among Honey Bees. Genome Res. 2021, 31, 1203–1215. [Google Scholar] [CrossRef] [PubMed]
- Schartl, M.; Kneitz, S.; Volkoff, H.; Adolfi, M.; Schmidt, C.; Fischer, P.; Minx, P.; Tomlinson, C.; Meyer, A.; Warren, W.C. The Piranha Genome Provides Molecular Insight Associated to Its Unique Feeding Behavior. Genome Biol. Evol. 2019, 11, 2099–2106. [Google Scholar] [CrossRef] [PubMed]
- Elsik, C.G.; Worley, K.C.; Bennett, A.K.; Beye, M.; Camara, F.; Childers, C.P.; de Graaf, D.C.; Debyser, G.; Deng, J.; Devreese, B.; et al. Finding the Missing Honey Bee Genes: Lessons Learned from a Genome Upgrade. BMC Genom. 2014, 15, 86. [Google Scholar] [CrossRef] [PubMed]
- Song, J.L.; Stoeckius, M.; Maaskola, J.; Friedländer, M.; Stepicheva, N.; Juliano, C.; Lebedeva, S.; Thompson, W.; Rajewsky, N.; Wessel, G.M. Select microRNAs Are Essential for Early Development in the Sea Urchin. Dev. Biol. 2012, 362, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Santos, D.; Feng, M.; Kolliopoulou, A.; Taning, C.N.T.; Sun, J.; Swevers, L. What Are the Functional Roles of Piwi Proteins and piRNAs in Insects? Insects 2023, 14, 187. [Google Scholar] [CrossRef]
- Lukic, S.; Nicolas, J.-C.; Levine, A.J. The Diversity of Zinc-Finger Genes on Human Chromosome 19 Provides an Evolutionary Mechanism for Defense against Inherited Endogenous Retroviruses. Cell Death Differ. 2014, 21, 381–387. [Google Scholar] [CrossRef]
- Baumgartner, L.; Handler, D.; Platzer, S.W.; Yu, C.; Duchek, P.; Brennecke, J. The Drosophila ZAD Zinc Finger Protein Kipferl Guides Rhino to piRNA Clusters. eLife 2022, 11, e80067. [Google Scholar] [CrossRef]
- Catlin, N.S.; Josephs, E.B. The Important Contribution of Transposable Elements to Phenotypic Variation and Evolution. Curr. Opin. Plant Biol. 2022, 65, 102140. [Google Scholar] [CrossRef]
- Wells, J.N.; Chang, N.-C.; McCormick, J.; Coleman, C.; Ramos, N.; Jin, B.; Feschotte, C. Transposable Elements Drive the Evolution of Metazoan Zinc Finger Genes. Genome Res. 2023, 33, 1325–1339. [Google Scholar] [CrossRef]
- Harrison, M.C.; Jongepier, E.; Robertson, H.M.; Arning, N.; Bitard-Feildel, T.; Chao, H.; Childers, C.P.; Dinh, H.; Doddapaneni, H.; Dugan, S.; et al. Hemimetabolous Genomes Reveal Molecular Basis of Termite Eusociality. Nat. Ecol. Evol. 2018, 2, 557–566. [Google Scholar] [CrossRef]
- Gebrie, A. Transposable Elements as Essential Elements in the Control of Gene Expression. Mob. DNA 2023, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, V.; Wysocka, J. Transposable Elements as a Potent Source of Diverse Cis-Regulatory Sequences in Mammalian Genomes. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190347. [Google Scholar] [CrossRef] [PubMed]
- Alser, M.; Rotman, J.; Deshpande, D.; Taraszka, K.; Shi, H.; Baykal, P.I.; Yang, H.T.; Xue, V.; Knyazev, S.; Singer, B.D.; et al. Technology Dictates Algorithms: Recent Developments in Read Alignment. Genome Biol. 2021, 22, 249. [Google Scholar] [CrossRef] [PubMed]
- van der Velde, A.; Fan, K.; Tsuji, J.; Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Weng, Z. Annotation of Chromatin States in 66 Complete Mouse Epigenomes during Development. Commun. Biol. 2021, 4, 239. [Google Scholar] [CrossRef]
- Brown, E.J.; Bachtrog, D. The Chromatin Landscape of Drosophila: Comparisons between Species, Sexes, and Chromosomes. Genome Res. 2014, 24, 1125–1137. [Google Scholar] [CrossRef]
- He, J.; Babarinde, I.A.; Sun, L.; Xu, S.; Chen, R.; Shi, J.; Wei, Y.; Li, Y.; Ma, G.; Zhuang, Q.; et al. Identifying Transposable Element Expression Dynamics and Heterogeneity during Development at the Single-Cell Level with a Processing Pipeline scTE. Nat. Commun. 2021, 12, 1456. [Google Scholar] [CrossRef]
- Treiber, C.D.; Waddell, S. Transposon Expression in the Drosophila Brain Is Driven by Neighboring Genes and Diversifies the Neural Transcriptome. Genome Res. 2020, 30, 1559–1569. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panyushev, N.; Selitskiy, M.; Melnichenko, V.; Lebedev, E.; Okorokova, L.; Adonin, L. Dynamic Evolution of Repetitive Elements and Chromatin States in Apis mellifera Subspecies. Genes 2024, 15, 89. https://doi.org/10.3390/genes15010089
Panyushev N, Selitskiy M, Melnichenko V, Lebedev E, Okorokova L, Adonin L. Dynamic Evolution of Repetitive Elements and Chromatin States in Apis mellifera Subspecies. Genes. 2024; 15(1):89. https://doi.org/10.3390/genes15010089
Chicago/Turabian StylePanyushev, Nick, Max Selitskiy, Vasilina Melnichenko, Egor Lebedev, Larisa Okorokova, and Leonid Adonin. 2024. "Dynamic Evolution of Repetitive Elements and Chromatin States in Apis mellifera Subspecies" Genes 15, no. 1: 89. https://doi.org/10.3390/genes15010089
APA StylePanyushev, N., Selitskiy, M., Melnichenko, V., Lebedev, E., Okorokova, L., & Adonin, L. (2024). Dynamic Evolution of Repetitive Elements and Chromatin States in Apis mellifera Subspecies. Genes, 15(1), 89. https://doi.org/10.3390/genes15010089