
The Microphthalmia-Associated Transcription Factor (MITF) and Its Role in the Structure and Function of the Eye

Abstract
:1. Introduction
2. The Role of Mitf in the RPE
Mitf and Ion Transport across the RPE
3. Mitf and the Vasculature of the Eye

{kind=link}
{kind=link}
| Genotype | Phenotype | Source |
|---|---|---|
| Mitfmi | White coat; eyes small and red; osteopetrosis; inner defects; incisors fail to erupt; deficiency of mast cells. | [7] |
| Mitfmi-rw | Colored marks around the neck; eyes small and red. | [75] |
| Mitfmi-bw | White coat with colored spots on rump and head; eyes small and red. | [76] |
| MitfMi-wh | White coat; eyes slightly pigmented and small. | [19] |
| Mitfmi-vga9 | White coat; eyes small and red. | [7] |
| Mitfmi-vit | Initial markings on the thorax and abdomen; gradual loss of pigmentation in coat and eye; defective RPE–photoreceptor interactions. | [77,78,79] |
| MitfMi-or | White coat; eyes small and red; osteopetrosis; incisors fail to erupt. | [80,81] |
| MitfMi-H | White coat; eyelids are closed at birth. | [82] |
| MitfMi-b | White coat; reduced eye pigmentation. | [83] |
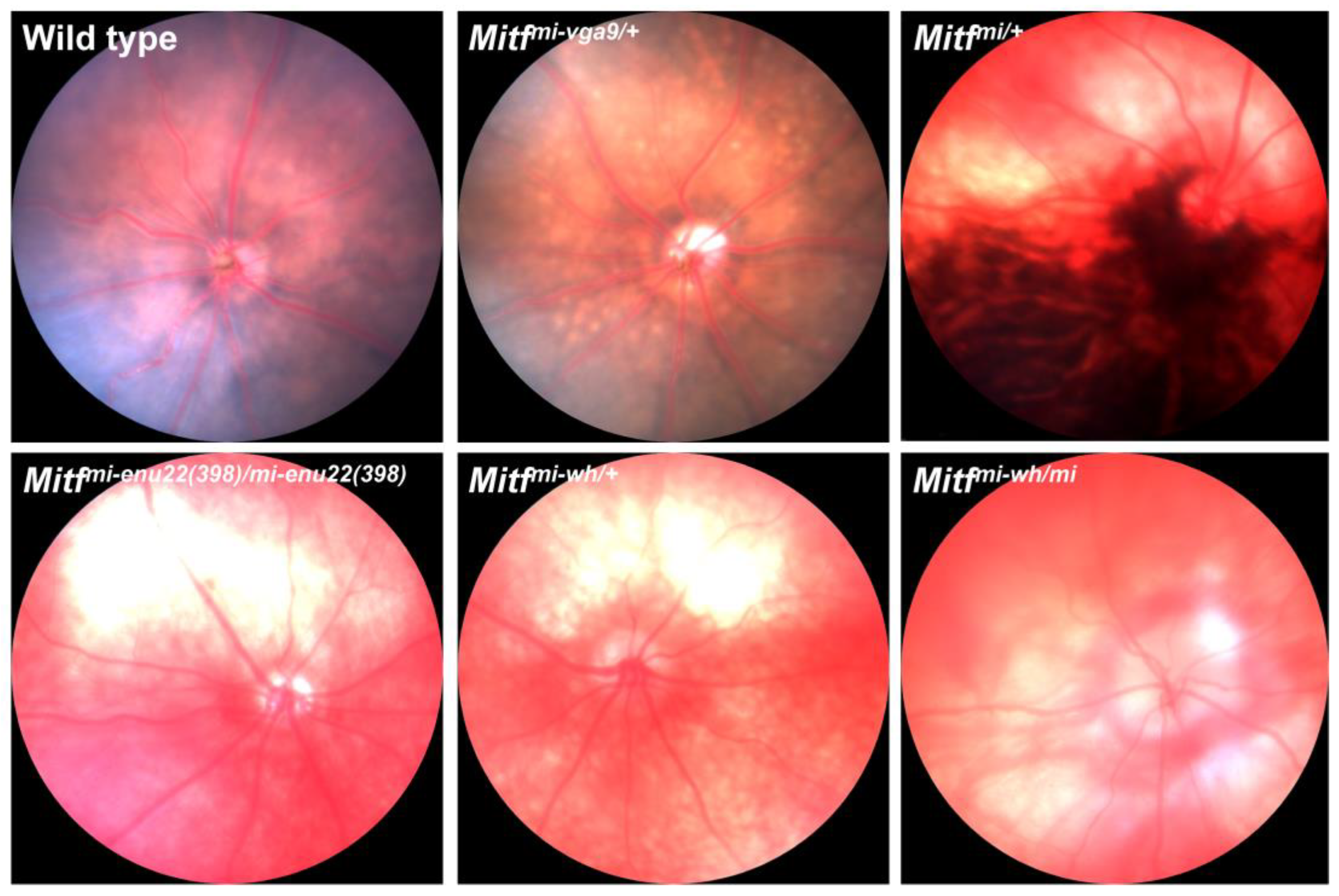
4. Mitf Mutations and Microphthalmia
5. Mitf and Postnatal Retinal Degeneration
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hertwig, P. Neue mutationen und koppelungsgruppen bei der hausmaus. Z. Indukt. Abstamm. Vererbungslehre 1942, 80, 220–246. [Google Scholar] [CrossRef]
- Gruneberg, H. Some observations on the microphthalmia gene in the mouse. J. Genet. 1948, 49, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Hara, Y.; Vyas, D.; Hodgkinson, C.; Fex, J.; Grundfast, K.; Arnheiter, H. Cochlear disorder associated with melanocyte anomaly in mice with a transgenic insertional mutation. Mol. Cell Neurosci. 1992, 3, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Hemesath, T.J.; Steingrimsson, E.; McGill, G.; Hansen, M.J.; Vaught, J.; Hodgkinson, C.A.; Arnheiter, H.; Copeland, N.G.; Jenkins, N.A.; Fisher, D.E. Microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes. Dev. 1994, 8, 2770–2780. [Google Scholar] [CrossRef]
- Steingrimsson, E.; Tessarollo, L.; Pathak, B.; Hou, L.; Arnheiter, H.; Copeland, N.G.; Jenkins, N.A. Mitf and Tfe3, two members of the Mitf-Tfe family of bHLH-Zip transcription factors, have important but functionally redundant roles in osteoclast development. Proc. Natl. Acad. Sci. USA 2002, 99, 4477–4482. [Google Scholar] [CrossRef]
- Goding, C.R. Mitf from neural crest to melanoma: Signal transduction and transcription in the melanocyte lineage. Genes. Dev. 2000, 14, 1712–1728. [Google Scholar] [CrossRef]
- Hodgkinson, C.A.; Moore, K.J.; Nakayama, A.; Steingrímsson, E.; Copeland, N.G.; Jenkins, N.A.; Arnheiter, H. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell 1993, 74, 395–404. [Google Scholar] [CrossRef]
- Nakayama, A.; Nguyen, M.T.T.; Chen, C.C.; Opdecamp, K.; Hodgkinson, C.A.; Amheiter, H. Mutations in microphthalmia, the mouse homolog of the human deafness gene MITF, affect neuroepithelial and neural crest-derived melanocytes differently. Mech. Dev. 1998, 70, 155–166. [Google Scholar] [CrossRef]
- Arnheiter, H. The discovery of the microphthalmia locus and its gene, Mitf. Pigment. Cell Melanoma Res. 2010, 23, 729–735. [Google Scholar] [CrossRef]
- Lu, S.Y.; Li, M.; Lin, Y.L. Mitf regulates osteoclastogenesis by modulating NFATc1 activity. Exp. Cell Res. 2014, 328, 32–43. [Google Scholar] [CrossRef]
- Ma, X.; Li, H.; Chen, Y.; Yang, J.; Chen, H.; Arnheiter, H.; Hou, L. The transcription factor MITF in RPE function and dysfunction. Prog. Retin. Eye Res. 2019, 73, 100766. [Google Scholar] [CrossRef] [PubMed]
- Tshori, S.; Gilon, D.; Beeri, R.; Nechushtan, H.; Kaluzhny, D.; Pikarsky, E.; Razin, E. Transcription factor MITF regulates cardiac growth and hypertrophy. J. Clin. Invest. 2006, 116, 2673–2681. [Google Scholar] [CrossRef] [PubMed]
- Bharti, K.; Nguyen, M.T.; Skuntz, S.; Bertuzzi, S.; Arnheiter, H. The other pigment cell: Specification and development of the pigmented epithelium of the vertebrate eye. Pigment. Cell Res. 2006, 19, 380–394. [Google Scholar] [CrossRef] [PubMed]
- Bumsted, K.M.; Barnstable, C.J. Dorsal retinal pigment epithelium differentiates as neural retina in the microphthalmia (mi/mi) mouse. Invest. Ophthalmol. Vis. Sci. 2000, 41, 903–908. [Google Scholar]
- Nguyen, M.; Arnheiter, H. Signaling and transcriptional regulation in early mammalian eye development: A link between FGF and MITF. Development 2000, 127, 3581–3591. [Google Scholar] [CrossRef]
- García-Llorca, A.; Aspelund, S.G.; Ogmundsdottir, M.H.; Steingrimsson, E.; Eysteinsson, T. The microphthalmia-associated transcription factor (Mitf) gene and its role in regulating eye function. Sci. Rep. 2019, 9, 15386. [Google Scholar] [CrossRef]
- García-Llorca, A.; Ólafsson, K.H.; Sigurdsson, A.T.; Eysteinsson, T. Progressive Cone-Rod Dystrophy and RPE Dysfunction in Mitf(mi/+) Mice. Genes 2023, 14, 1458. [Google Scholar] [CrossRef]
- Steingrímsson, E.; Copeland, N.G.; Jenkins, N.A. Melanocytes and the microphthalmia transcription factor network. Annu. Rev. Genet. 2004, 38, 365–411. [Google Scholar] [CrossRef]
- Steingrímsson, E.; Moore, K.J.; Lamoreux, M.L.; Ferré-D’Amaré, A.R.; Burley, S.K.; Zimring, D.C.; Skow, L.C.; Hodgkinson, C.A.; Arnheiter, H.; Copeland, N.G.; et al. Molecular basis of mouse microphthalmia (mi) mutations helps explain their developmental and phenotypic consequences. Nat. Genet. 1994, 8, 256–263. [Google Scholar] [CrossRef]
- Yasumoto, K.; Amae, S.; Udono, T.; Fuse, N.; Takeda, K.; Shibahara, S. A big gene linked to small eyes encodes multiple Mitf isoforms: Many promoters make light work. Pigment. Cell Res. 1998, 11, 329–336. [Google Scholar] [CrossRef]
- Read, A.P.; Newton, V.E. Waardenburg syndrome. J. Med. Genet. 1997, 34, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Amiel, J.; Watkin, P.M.; Tassabehji, M.; Read, A.P.; Winter, R.M. Mutation of the MITF gene in albinism-deafness syndrome (Tietz syndrome). Clin. Dysmorphol. 1998, 7, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.D.; Kelley, P.M.; Kenyon, J.B.; Hoover, D. Tietz syndrome (hypopigmentation/deafness) caused by mutation of MITF. J. Med. Genet. 2000, 37, 446–448. [Google Scholar] [CrossRef]
- George, A.; Zand, D.J.; Hufnagel, R.B.; Sharma, R.; Sergeev, Y.V.; Legare, J.M.; Rice, G.M.; Scott Schwoerer, J.A.; Rius, M.; Tetri, L.; et al. Biallelic Mutations in MITF Cause Coloboma, Osteopetrosis, Microphthalmia, Macrocephaly, Albinism, and Deafness. Am. J. Hum. Genet. 2016, 99, 1388–1394. [Google Scholar] [CrossRef]
- Bertolotto, C.; Lesueur, F.; Giuliano, S.; Strub, T.; de Lichy, M.; Bille, K.; Dessen, P.; d’Hayer, B.; Mohamdi, H.; Remenieras, A.; et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 2011, 480, 94–98. [Google Scholar] [CrossRef]
- Murakami, H.; Arnheiter, H. Sumoylation modulates transcriptional activity of MITF in a promoter-specific manner. Pigment. Cell Res. 2005, 18, 265–277. [Google Scholar] [CrossRef]
- Yokoyama, S.; Woods, S.L.; Boyle, G.M.; Aoude, L.G.; MacGregor, S.; Zismann, V.; Gartside, M.; Cust, A.E.; Haq, R.; Harland, M.; et al. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature 2011, 480, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed]
- Wimmers, S.; Karl, M.O.; Strauss, O. Ion channels in the RPE. Prog. Retin. Eye Res. 2007, 26, 263–301. [Google Scholar] [CrossRef]
- Weiter, J.J.; Delori, F.C.; Wing, G.L.; Fitch, K.A. Retinal pigment epithelial lipofuscin and melanin and choroidal melanin in human eyes. Invest. Ophthalmol. Vis. Sci. 1986, 27, 145–152. [Google Scholar]
- Cheli, Y.; Ohanna, M.; Ballotti, R.; Bertolotto, C. Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment. Cell Melanoma Res. 2010, 23, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.J.; Shao, J.; Smutko, J.S.; Dussault, B.J.; Nagle, D.L.; Woolf, E.A.; Holmgren, L.M.; Moore, K.J.; Shyjan, A.W. Chromosomal localization and genomic characterization of the mouse melastatin gene (Mlsn1). Genomics 1998, 54, 116–123. [Google Scholar] [CrossRef]
- Lu, S.; Slominski, A.; Yang, S.E.; Sheehan, C.; Ross, J.; Carlson, J.A. The correlation of TRPM1 (Melastatin) mRNA expression with microphthalmia-associated transcription factor (MITF) and other melanogenesis-related proteins in normal and pathological skin, hair follicles and melanocytic nevi. J. Cutan. Pathol. 2010, 37, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Krizaj, D.; Cordeiro, S.; Strauss, O. Retinal TRP channels: Cell-type-specific regulators of retinal homeostasis and multimodal integration. Prog. Retin. Eye Res. 2023, 92, 101114. [Google Scholar] [CrossRef] [PubMed]
- Gomez, N.M.; Lu, W.; Lim, J.C.; Kiselyov, K.; Campagno, K.E.; Grishchuk, Y.; Slaugenhaupt, S.A.; Pfeffer, B.A.; Fliesler, S.J.; Mitchell, C.H. Robust lysosomal calcium signaling through channel TRPML1 is impaired by lysosomal lipid accumulation. FASEB J. 2018, 32, 782–794. [Google Scholar] [CrossRef]
- Beckel, J.M.; Gomez, N.M.; Lu, W.; Campagno, K.E.; Nabet, B.; Albalawi, F.; Lim, J.C.; Boesze-Battaglia, K.; Mitchell, C.H. Stimulation of TLR3 triggers release of lysosomal ATP in astrocytes and epithelial cells that requires TRPML1 channels. Sci. Rep. 2018, 8, 5726. [Google Scholar] [CrossRef]
- Adijanto, J.; Castorino, J.J.; Wang, Z.X.; Maminishkis, A.; Grunwald, G.B.; Philp, N.J. Microphthalmia-associated transcription factor (MITF) promotes differentiation of human retinal pigment epithelium (RPE) by regulating microRNAs-204/211 expression. J. Biol. Chem. 2012, 287, 20491–20503. [Google Scholar] [CrossRef]
- Du, S.W.; Komirisetty, R.; Lewandowski, D.; Choi, E.H.; Panas, D.; Suh, S.; Tabaka, M.; Radu, R.A.; Palczewski, K. Conditional deletion of miR-204 and miR-211 in murine retinal pigment epithelium results in retinal degeneration. J. Biol. Chem. 2024, 300, 107344. [Google Scholar] [CrossRef]
- Gilliam, J.C.; Wensel, T.G. TRP channel gene expression in the mouse retina. Vis. Vision. Res. 2011, 51, 2440–2452. [Google Scholar] [CrossRef]
- Morgans, C.W.; Zhang, J.; Jeffrey, B.G.; Nelson, S.M.; Burke, N.S.; Duvoisin, R.M.; Brown, R.L. TRPM1 is required for the depolarizing light response in retinal ON-bipolar cells. Proc. Natl. Acad. Sci. USA 2009, 106, 19174–19178. [Google Scholar] [CrossRef]
- Shen, Y.; Heimel, J.A.; Kamermans, M.; Peachey, N.S.; Gregg, R.G.; Nawy, S. A transient receptor potential-like channel mediates synaptic transmission in rod bipolar cells. J. Neurosci. 2009, 29, 6088–6093. [Google Scholar] [CrossRef]
- Xu, Y.; Dhingra, A.; Fina, M.E.; Koike, C.; Furukawa, T.; Vardi, N. MGluR6 deletion renders the TRPM1 channel in retina inactive. J. Neurophysiol. 2012, 107, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Schneider, F.M.; Mohr, F.; Behrendt, M.; Oberwinkler, J. Properties and functions of TRPM1 channels in the dendritic tips of retinal ON-bipolar cells. Eur. J. Cell Biol. 2015, 94, 420–427. [Google Scholar] [CrossRef]
- Peachey, N.S.; Pearring, J.N.; Bojang, P., Jr.; Hirschtritt, M.E.; Sturgill-Short, G.; Ray, T.A.; Furukawa, T.; Koike, C.; Goldberg, A.F.; Shen, Y.; et al. Depolarizing bipolar cell dysfunction due to a Trpm1 point mutation. J. Neurophysiol. 2012, 108, 2442–2451. [Google Scholar] [CrossRef]
- Audo, I.; Kohl, S.; Leroy, B.P.; Munier, F.L.; Guillonneau, X.; Mohand-Said, S.; Bujakowska, K.; Nandrot, E.F.; Lorenz, B.; Preising, M.; et al. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2009, 85, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Esumi, N.; Kachi, S.; Campochiaro, P.A.; Zack, D.J. VMD2 promoter requires two proximal E-box sites for its activity in vivo and is regulated by the MITF-TFE family. J. Biol. Chem. 2007, 282, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Esumi, N.; Kachi, S.; Hackler, L., Jr.; Masuda, T.; Yang, Z.; Campochiaro, P.A.; Zack, D.J. BEST1 expression in the retinal pigment epithelium is modulated by OTX family members. Hum. Mol. Genet. 2009, 18, 128–141. [Google Scholar] [CrossRef]
- Masuda, T.; Esumi, N. SOX9, through interaction with microphthalmia-associated transcription factor (MITF) and OTX2, regulates BEST1 expression in the retinal pigment epithelium. J. Biol. Chem. 2010, 285, 26933–26944. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, A.D.; Marmorstein, L.Y.; Rayborn, M.; Wang, X.; Hollyfield, J.G.; Petrukhin, K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2000, 97, 12758–12763. [Google Scholar] [CrossRef]
- Martínez-Morales, J.R.; Dolez, V.; Rodrigo, I.; Zaccarini, R.; Leconte, L.; Bovolenta, P.; Saule, S. OTX2 activates the molecular network underlying retina pigment epithelium differentiation. J. Biol. Chem. 2003, 278, 21721–21731. [Google Scholar] [CrossRef]
- Poche, R.A.; Furuta, Y.; Chaboissier, M.C.; Schedl, A.; Behringer, R.R. Sox9 is expressed in mouse multipotent retinal progenitor cells and functions in Muller glial cell development. J. Comp. Neurol. 2008, 510, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Tsunenari, T.; Yau, K.W.; Nathans, J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc. Natl. Acad. Sci. USA 2002, 99, 4008–4013. [Google Scholar] [CrossRef]
- Rosenthal, R.; Bakall, B.; Kinnick, T.; Peachey, N.; Wimmers, S.; Wadelius, C.; Marmorstein, A.; Strauss, O. Expression of bestrophin-1, the product of the VMD2 gene, modulates voltage-dependent Ca2+ channels in retinal pigment epithelial cells. FASEB J. 2006, 20, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; Guziewicz, K.E.; Lee, C.J.; Kalathur, R.C.; Pulido, J.S.; Marmorstein, L.Y.; Marmorstein, A.D. Bestrophin 1 and retinal disease. Prog. Retin. Eye Res. 2017, 58, 45–69. [Google Scholar] [CrossRef]
- Marmorstein, A.D.; Kinnick, T.R.; Stanton, J.B.; Johnson, A.A.; Lynch, R.M.; Marmorstein, L.Y. Bestrophin-1 influences transepithelial electrical properties and Ca2+ signaling in human retinal pigment epithelium. Mol. Vis. 2015, 21, 347–359. [Google Scholar]
- Marmorstein, L.Y.; Wu, J.; McLaughlin, P.; Yocom, J.; Karl, M.O.; Neussert, R.; Wimmers, S.; Stanton, J.B.; Gregg, R.G.; Strauss, O.; et al. The light peak of the electroretinogram is dependent on voltage-gated calcium channels and antagonized by bestrophin (best-1). J. Gen. Physiol. 2006, 127, 577–589. [Google Scholar] [CrossRef]
- Chowers, I.; Tiosano, L.; Audo, I.; Grunin, M.; Boon, C.J. Adult-onset foveomacular vitelliform dystrophy: A fresh perspective. Prog. Retin. Eye Res. 2015, 47, 64–85. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.; Eysteinsson, T.; Steingrimsson, E. Electroretinographic assessment of retinal function in microphthalmia mutant mice. Exp. Eye Res. 2004, 78, 837–848. [Google Scholar] [CrossRef]
- Saint-Geniez, M.; Kurihara, T.; Sekiyama, E.; Maldonado, A.E.; D’Amore, P.A. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc. Natl. Acad. Sci. USA 2009, 106, 18751–18756. [Google Scholar] [CrossRef]
- Sakagami, K.; Kodama, T.; Puro, D.G. PDGF-induced coupling of function with metabolism in microvascular pericytes of the retina. Invest. Ophthalmol. Vis. Sci. 2001, 42, 1939–1944. [Google Scholar]
- Rousseau, B.; Larrieu-Lahargue, F.; Bikfalvi, A.; Javerzat, S. Involvement of fibroblast growth factors in choroidal angiogenesis and retinal vascularization. Exp. Eye Res. 2003, 77, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.; Schlunck, G.; Hansen, L.L.; Agostini, H.T. Differential expression of angioregulatory factors in normal and CNV-derived human retinal pigment epithelium. Graefes Arch. Clin. Exp. Ophthalmol. 2004, 242, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Murugeswari, P.; Subramani, M.; Jayadev, C.; Shetty, R.; Das, D. Retinal pigment epithelium-secretome: A diabetic retinopathy perspective. Cytokine 2017, 95, 126–135. [Google Scholar] [CrossRef]
- Ma, X.; Pan, L.; Jin, X.; Dai, X.; Li, H.; Wen, B.; Chen, Y.; Ma, A.; Qu, J.; Hou, L. Microphthalmia-associated transcription factor acts through PEDF to regulate RPE cell migration. Exp. Cell Res. 2012, 318, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Saint-Geniez, M.; Maldonado, A.E.; D’Amore, P.A. VEGF expression and receptor activation in the choroid during development and in the adult. Invest. Ophthalmol. Vis. Sci. 2006, 47, 3135–3142. [Google Scholar] [CrossRef]
- Ford, K.M.; D’Amore, P.A. Molecular regulation of vascular endothelial growth factor expression in the retinal pigment epithelium. Mol. Vis. 2012, 18, 519–527. [Google Scholar]
- Marneros, A.G.; Fan, J.; Yokoyama, Y.; Gerber, H.P.; Ferrara, N.; Crouch, R.K.; Olsen, B.R. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am. J. Pathol. 2005, 167, 1451–1459. [Google Scholar] [CrossRef]
- Shibuya, H.; Watanabe, R.; Maeno, A.; Ichimura, K.; Tamura, M.; Wakana, S.; Shiroishi, T.; Ohba, K.; Takeda, K.; Tomita, H.; et al. Melanocytes contribute to the vasculature of the choroid. Genes. Genet. Syst. 2018, 93, 51–58. [Google Scholar] [CrossRef]
- Becerra, S.P.; Fariss, R.N.; Wu, Y.Q.; Montuenga, L.M.; Wong, P.; Pfeffer, B.A. Pigment epithelium-derived factor in the monkey retinal pigment epithelium and interphotoreceptor matrix: Apical secretion and distribution. Exp. Eye Res. 2004, 78, 223–234. [Google Scholar] [CrossRef]
- Karakousis, P.C.; John, S.K.; Behling, K.C.; Surace, E.M.; Smith, J.E.; Hendrickson, A.; Tang, W.X.; Bennett, J.; Milam, A.H. Localization of pigment epithelium derived factor (PEDF) in developing and adult human ocular tissues. Mol. Vis. 2001, 7, 154–163. [Google Scholar]
- Huang, Q.; Wang, S.; Sorenson, C.M.; Sheibani, N. PEDF-deficient mice exhibit an enhanced rate of retinal vascular expansion and are more sensitive to hyperoxia-mediated vessel obliteration. Exp. Eye Res. 2008, 87, 226–241. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, J.; Geng, H.; Li, L.; Li, J.; Cheng, B.; Ma, X.; Li, H.; Hou, L. Photoreceptor degeneration in microphthalmia (Mitf) mice: Partial rescue by pigment epithelium-derived factor. Dis. Model. Mech. 2019, 12, dmm035642. [Google Scholar] [CrossRef] [PubMed]
- Au—García-Llorca, A.; Au—Reynisson, H.; Au—Eysteinsson, T. Measuring Retinal Vessel Diameter from Mouse Fluorescent Angiography Images. J. Vis. Exp. 2023, 195, e64964. [Google Scholar] [CrossRef]
- Danielsson, S.B.; Garcia-Llorca, A.; Reynisson, H.; Eysteinsson, T. Mouse microphthalmia-associated transcription factor (Mitf) mutations affect the structure of the retinal vasculature. Acta Ophthalmol. 2022, 100, 911–918. [Google Scholar] [CrossRef]
- Bharti, K.; Liu, W.; Csermely, T.; Bertuzzi, S.; Arnheiter, H. Alternative promoter use in eye development: The complex role and regulation of the transcription factor MITF. Development 2008, 135, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Steingrímsson, E.; Arnheiter, H.; Hallsson, J.H.; Lamoreux, M.L.; Copeland, N.G.; Jenkins, N.A. Interallelic complementation at the mouse Mitf locus. Genetics 2003, 163, 267–276. [Google Scholar] [CrossRef]
- Lerner, A.B.; Shiohara, T.; Boissy, R.E.; Jacobson, K.A.; Lamoreux, M.L.; Moellmann, G.E. A mouse model for vitiligo. J. Invest. Dermatol. 1986, 87, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.B. C57BL/6J-vit/vit mouse model of retinal degeneration: Light microscopic analysis and evaluation of rhodopsin levels. Exp. Eye Res. 1992, 55, 903–910. [Google Scholar] [CrossRef]
- Smith, S.B.; Hamasaki, D.I. Electroretinographic study of the C57BL/6-mivit/mivit mouse model of retinal degeneration. Invest. Ophthalmol. Vis. Sci. 1994, 35, 3119–3123. [Google Scholar]
- Nii, A.; Steingrímsson, E.; Copeland, N.G.; Jenkins, N.A.; Ward, J.M. Mild osteopetrosis in the microphthalmia-oak ridge mouse. A model for intermediate autosomal recessive osteopetrosis in humans. Am. J. Pathol. 1995, 147, 1871–1882. [Google Scholar]
- Sharma, S.M.; Sif, S.; Ostrowski, M.C.; Sankar, U. Defective co-activator recruitment in osteoclasts from microphthalmia-oak ridge mutant mice. J. Cell Physiol. 2009, 220, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Thaung, C.; West, K.; Clark, B.J.; McKie, L.; Morgan, J.E.; Arnold, K.; Nolan, P.M.; Peters, J.; Hunter, A.J.; Brown, S.D.; et al. Novel ENU-induced eye mutations in the mouse: Models for human eye disease. Hum. Mol. Genet. 2002, 11, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Steingrímsson, E.; Nii, A.; Fisher, D.E.; Ferré-D’Amaré, A.R.; McCormick, R.J.; Russell, L.B.; Burley, S.K.; Ward, J.M.; Jenkins, N.A.; Copeland, N.G. The semidominant Mi(b) mutation identifies a role for the HLH domain in DNA binding in addition to its role in protein dimerization. Embo J. 1996, 15, 6280–6289. [Google Scholar] [CrossRef] [PubMed]
- Bumsted, K.M.; Rizzolo, L.J.; Barnstable, C.J. Defects in the MITF (mi/mi) apical surface are associated with a failure of outer segment elongation. Exp. Eye Res. 2001, 73, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Konyukhov, B.V.; Sazhina, M.V. Interaction of the genes of ocular retardation and microphthalmia in mice. Folia Biol. 1966, 12, 116–123. [Google Scholar]
- Konyukhov, B.V.; Osipov, V.V. Interallelic complementation of microphthalmia and white genes in mice. Genetika 1968, 4, 65–76. [Google Scholar]
- Steingrimsson, E. Interpretation of complex phenotypes: Lessons from the Mitf gene. Pigment. Cell Melanoma Res. 2010, 23, 736–740. [Google Scholar] [CrossRef]
- Justice, M.J.; Noveroske, J.K.; Weber, J.S.; Zheng, B.; Bradley, A. Mouse ENU mutagenesis. Hum. Mol. Genet. 1999, 8, 1955–1963. [Google Scholar] [CrossRef]
- Hansdottir, A.G.; Palsdottir, K.; Favor, J.; Neuhauser-Klaus, A.; Fuchs, H.; de Angelis, M.H.; Steingrimsson, E. The novel mouse microphthalmia mutations Mitfmi-enu5 and Mitfmi-bcc2 produce dominant negative Mitf proteins. Genomics 2004, 83, 932–935. [Google Scholar] [CrossRef]
- Hara, Y.; Battey, J.; Gainer, H. Structure of mouse vasopressin and oxytocin genes. Brain Res. Mol. Brain Res. 1990, 8, 319–324. [Google Scholar] [CrossRef]
- Tang, M.; Pawlyk, B.S.; Kosaras, B.; Berson, E.L.; Sidman, R.L. ERG abnormalities in relation to histopathologic findings in vitiligo mutant mice. Exp. Eye Res. 1997, 65, 215–222. [Google Scholar] [CrossRef]
- Boissy, R.E.; Moellmann, G.E.; Lerner, A.B. Morphology of melanocytes in hair bulbs and eyes of vitiligo mice. Am. J. Pathol. 1987, 127, 380–388. [Google Scholar] [PubMed]
- Sidman, R.L.; Kosaras, B.; Tang, M. Pigment epithelial and retinal phenotypes in the vitiligo mivit, mutant mouse. Invest. Ophthalmol. Vis. Sci. 1996, 37, 1097–1115. [Google Scholar] [PubMed]
- Chang, B.; Hawes, N.L.; Hurd, R.E.; Davisson, M.T.; Nusinowitz, S.; Heckenlively, J.R. Retinal degeneration mutants in the mouse. Vis. Vision. Res. 2002, 42, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.B.; Cope, B.K.; McCoy, J.R.; McCool, D.J.; Defoe, D.M. Reduction of phagosomes in the vitiligo (C57BL/6-mivit/mivit) mouse model of retinal degeneration. Invest. Ophthalmol. Vis. Sci. 1994, 35, 3625–3632. [Google Scholar]
- Nir, I.; Ransom, N.; Smith, S.B. Ultrastructural features of retinal dystrophy in mutant vitiligo mice. Exp. Eye Res. 1995, 61, 363–377. [Google Scholar] [CrossRef]
- Moore, K.J. Insight into the Microphthalmia Gene. Trends Genet. 1995, 11, 442–448. [Google Scholar] [CrossRef]
- Wolfe, H.G. New allele at the mi locus. Mouse News Let. 1962, 26, 35. [Google Scholar]
- Wolfe, H.G.; Coleman, D.l. Mi-spotted: A mutation in the mouse. Genet. Res. Camb. 1964, 5, 432–440. [Google Scholar] [CrossRef]
- Bertolotto, C.; Buscà, R.; Abbe, P.; Bille, K.; Aberdam, E.; Ortonne, J.P.; Ballotti, R. Different cis-acting elements are involved in the regulation of TRP1 and TRP2 promoter activities by cyclic AMP: Pivotal role of M boxes (GTCATGTGCT) and of microphthalmia. Mol. Cell Biol. 1998, 18, 694–702. [Google Scholar] [CrossRef]
- Zhang, H.; Luo, H.; Chen, H.; Mei, L.; He, C.; Jiang, L.; Li, J.D.; Feng, Y. Functional analysis of MITF gene mutations associated with Waardenburg syndrome type 2. FEBS Lett. 2012, 586, 4126–4131. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Hao, Z.; Luo, H.; He, C.; Mei, L.; Liu, Y.; Wang, X.; Niu, Z.; Chen, H.; Li, J.D.; et al. Functional analysis of a nonstop mutation in MITF gene identified in a patient with Waardenburg syndrome type 2. J. Hum. Genet. 2017, 62, 703–709. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Llorca, A.; Eysteinsson, T. The Microphthalmia-Associated Transcription Factor (MITF) and Its Role in the Structure and Function of the Eye. Genes 2024, 15, 1258. https://doi.org/10.3390/genes15101258
García-Llorca A, Eysteinsson T. The Microphthalmia-Associated Transcription Factor (MITF) and Its Role in the Structure and Function of the Eye. Genes. 2024; 15(10):1258. https://doi.org/10.3390/genes15101258
Chicago/Turabian StyleGarcía-Llorca, Andrea, and Thor Eysteinsson. 2024. "The Microphthalmia-Associated Transcription Factor (MITF) and Its Role in the Structure and Function of the Eye" Genes 15, no. 10: 1258. https://doi.org/10.3390/genes15101258