
Transcriptomic and Proteomic Analyses of the Immune Mechanism in Pathogenetic and Resistant Chinese Soft-Shelled Turtle (Pelodiscus sinensis) Infected with Aeromonas hydrophila

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Sample Collection
2.2. RNA Extraction and Construction of cDNA Libraries
2.3. Protein Extraction and LC-MS/MS Analysis
2.4. Bioinformatic Analysis
2.5. Correlation Analysis of Transcriptome and Proteome
2.6. Quantitative Real Time PCR (qRT-PCR)
3. Results
3.1. Symptoms in Chinese Soft-Shelled Turtles after A. hydrophila Infection
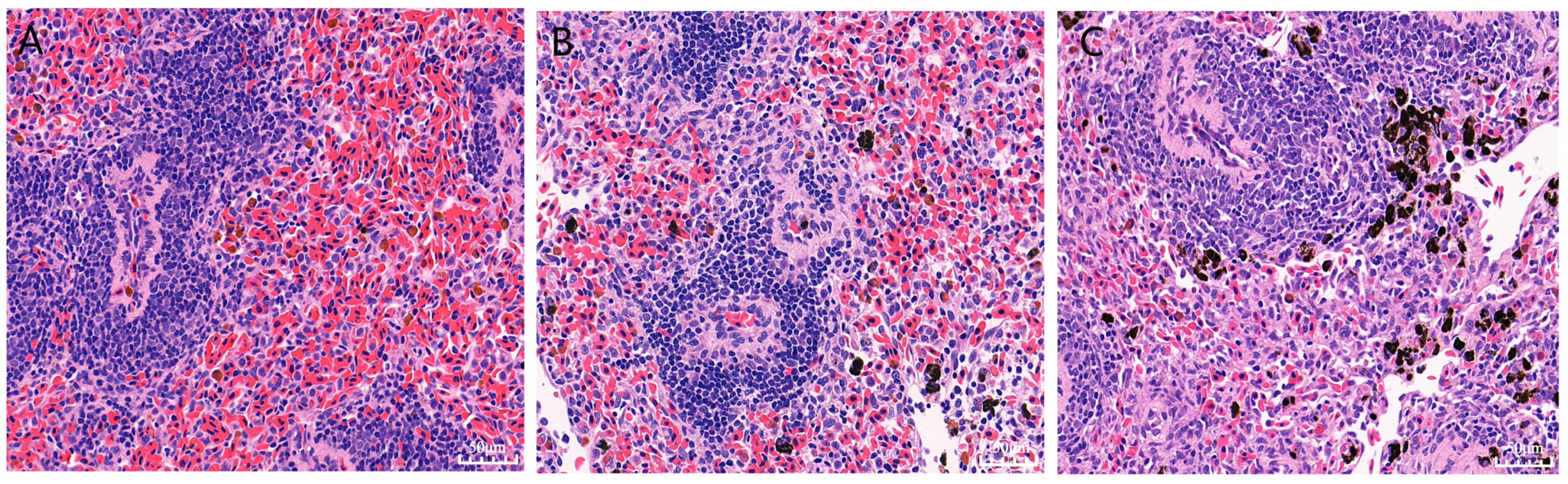
3.2. Pathological Analysis of the Chinese Soft-Shelled Turtles’ Spleens
3.3. Transcriptomic Analysis
3.3.1. Illumina Sequencing Results
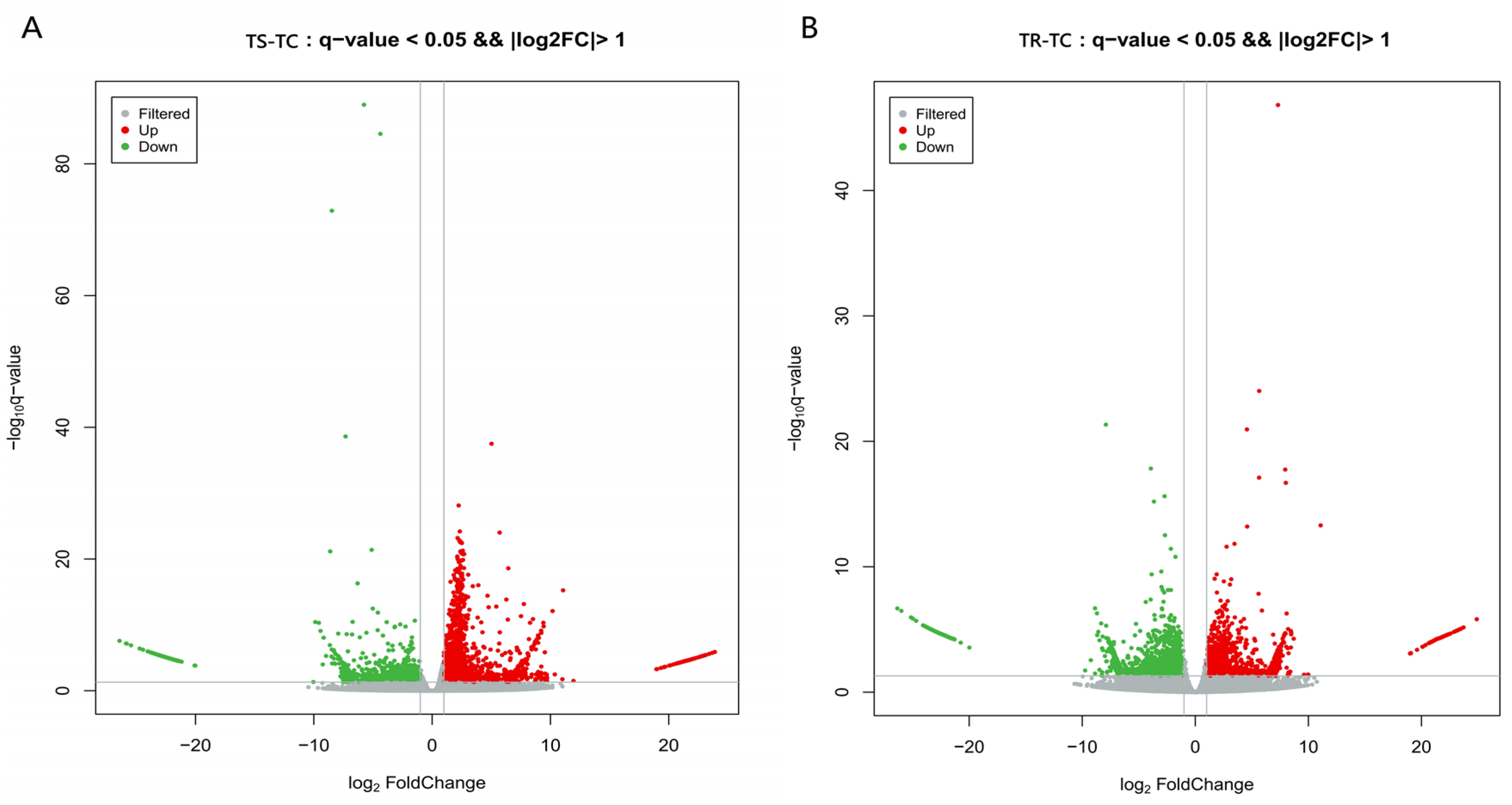
3.3.2. Identification of DEGs
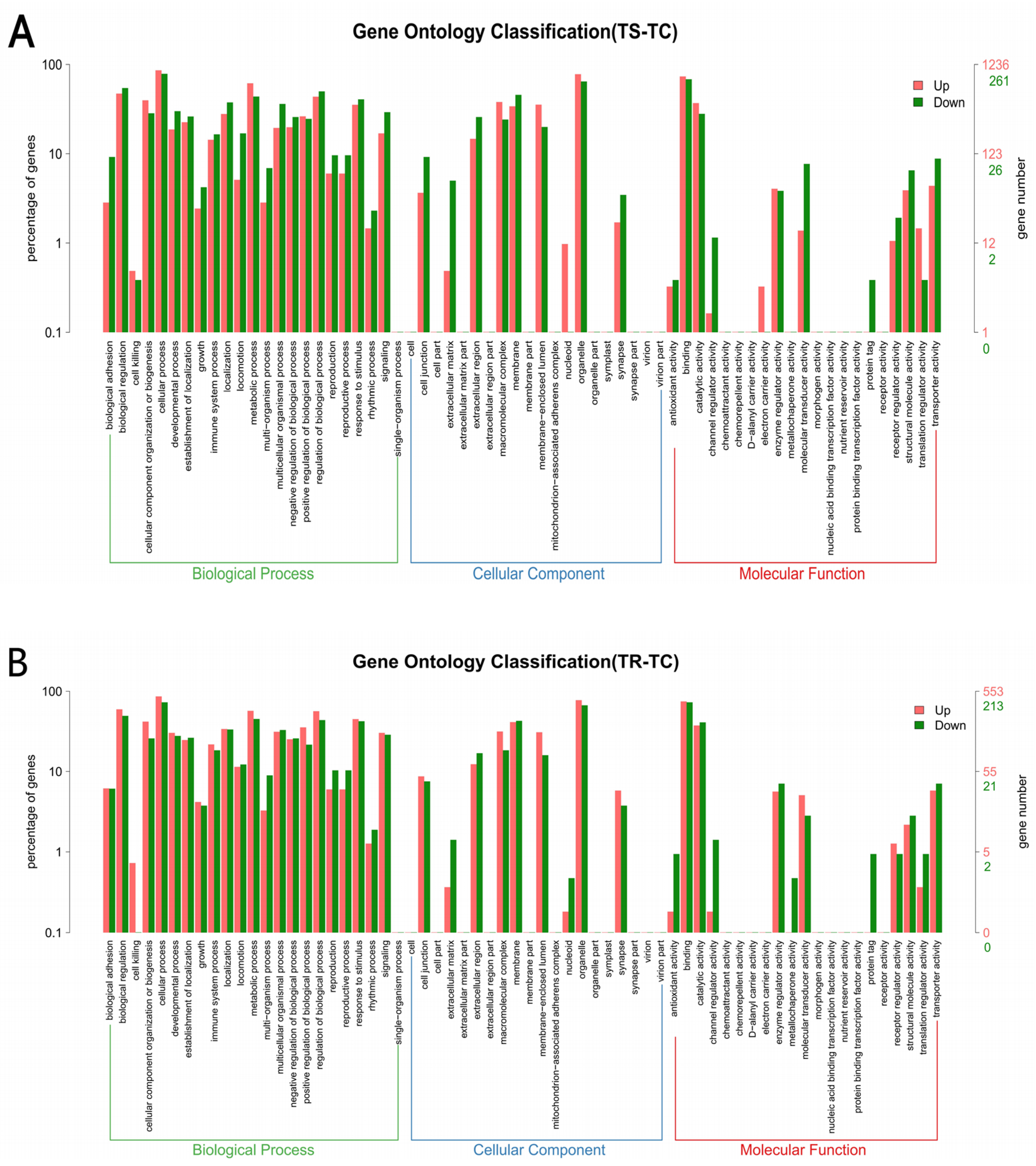
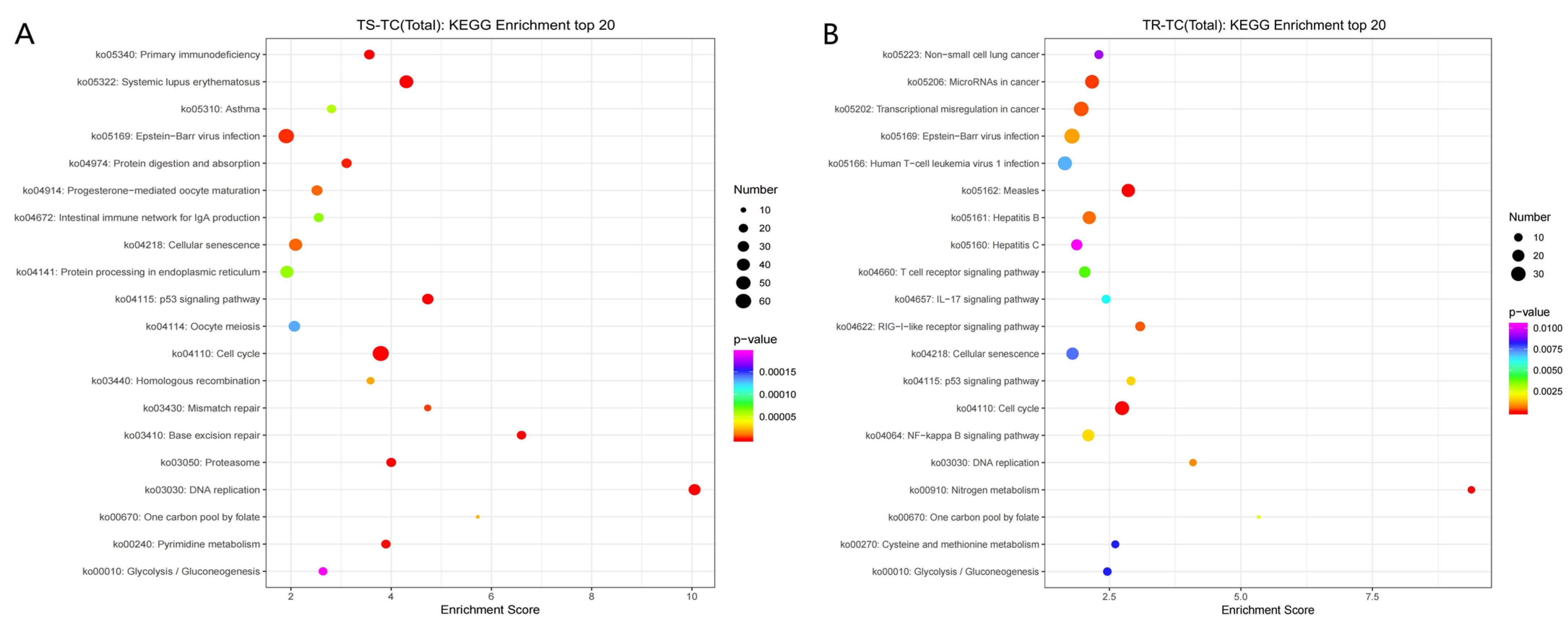
3.3.3. GO and KEGG Enrichment Analyses of DEGs
3.4. Proteomic Analysis
3.4.1. Identification of DEPs
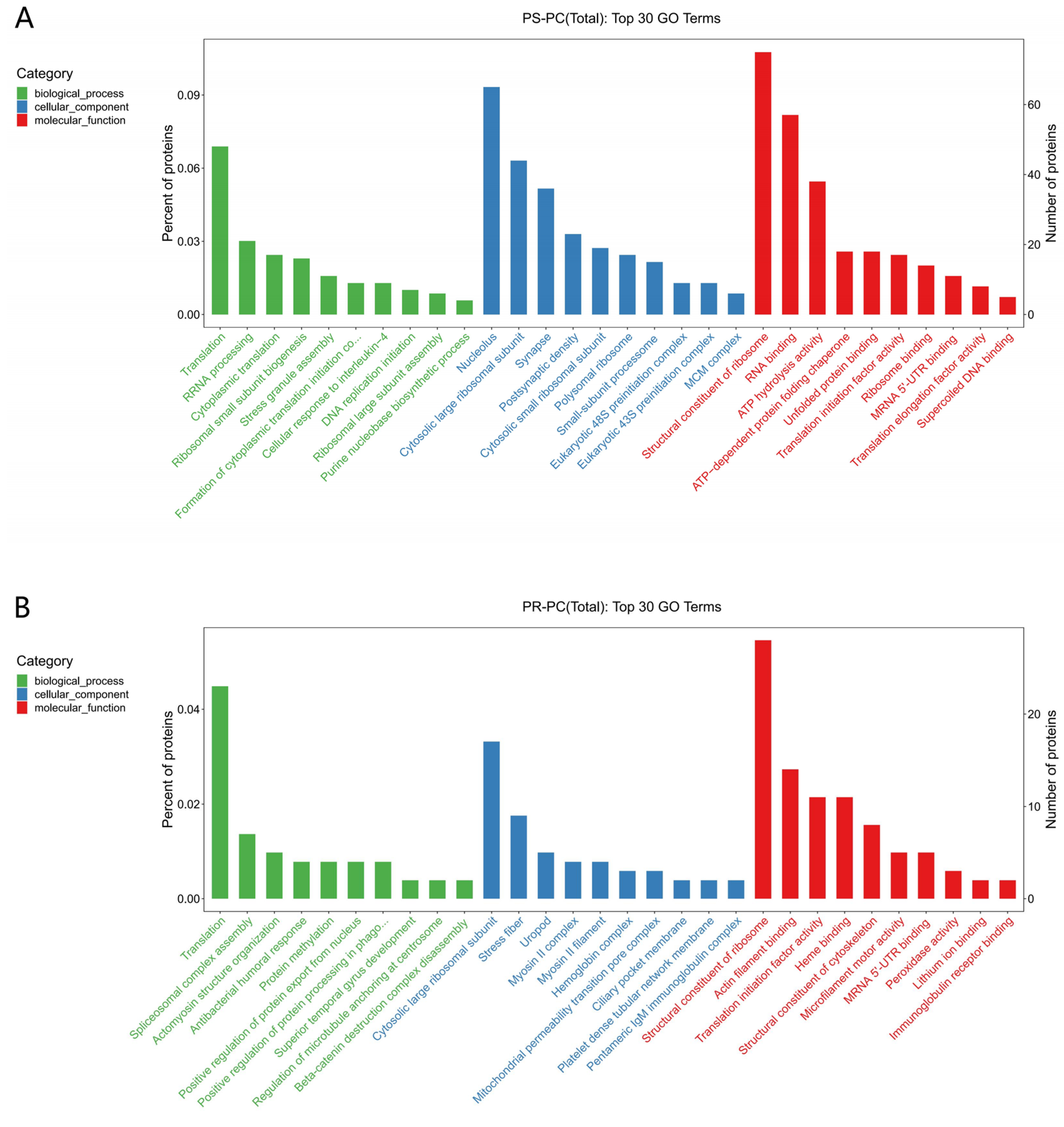
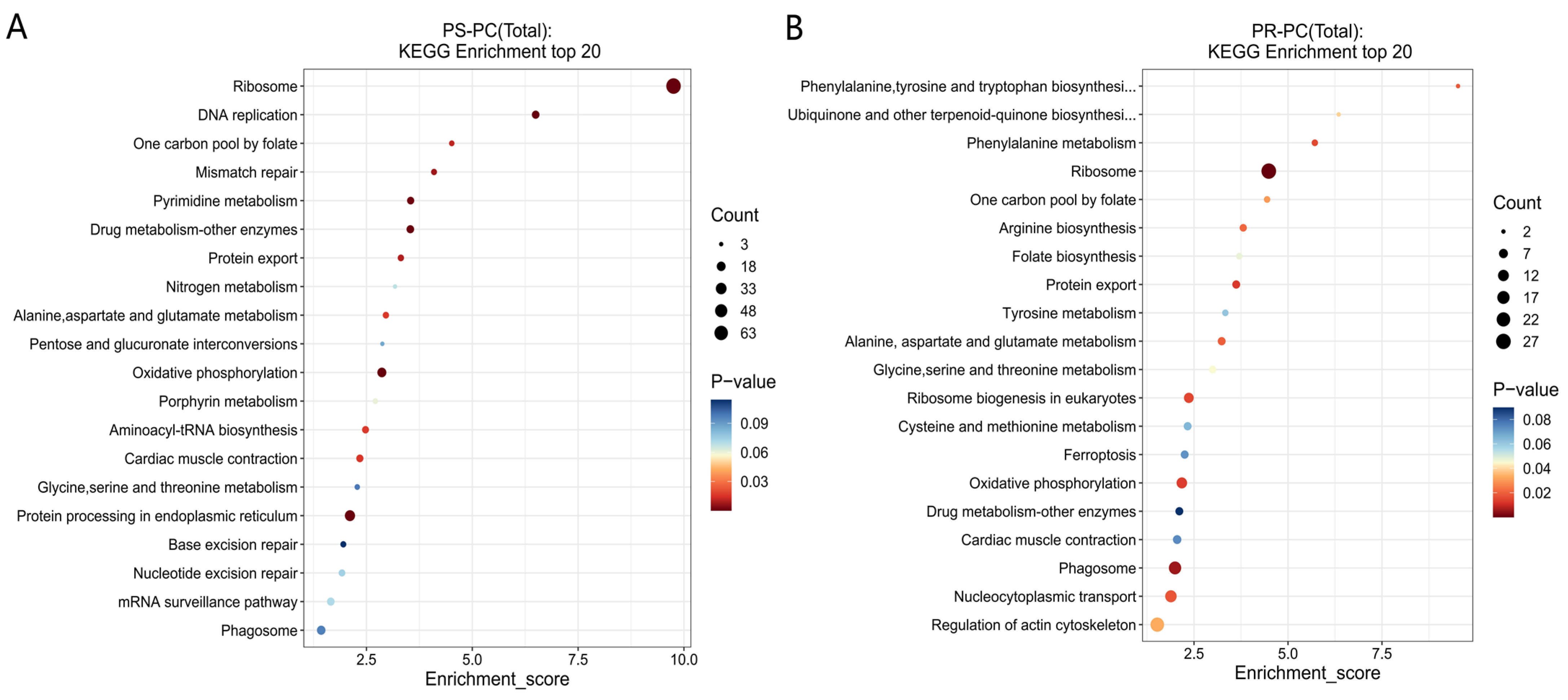
3.4.2. GO and KEGG Functional Analyses of DEPs
3.4.3. Differential Protein Interaction Analysis
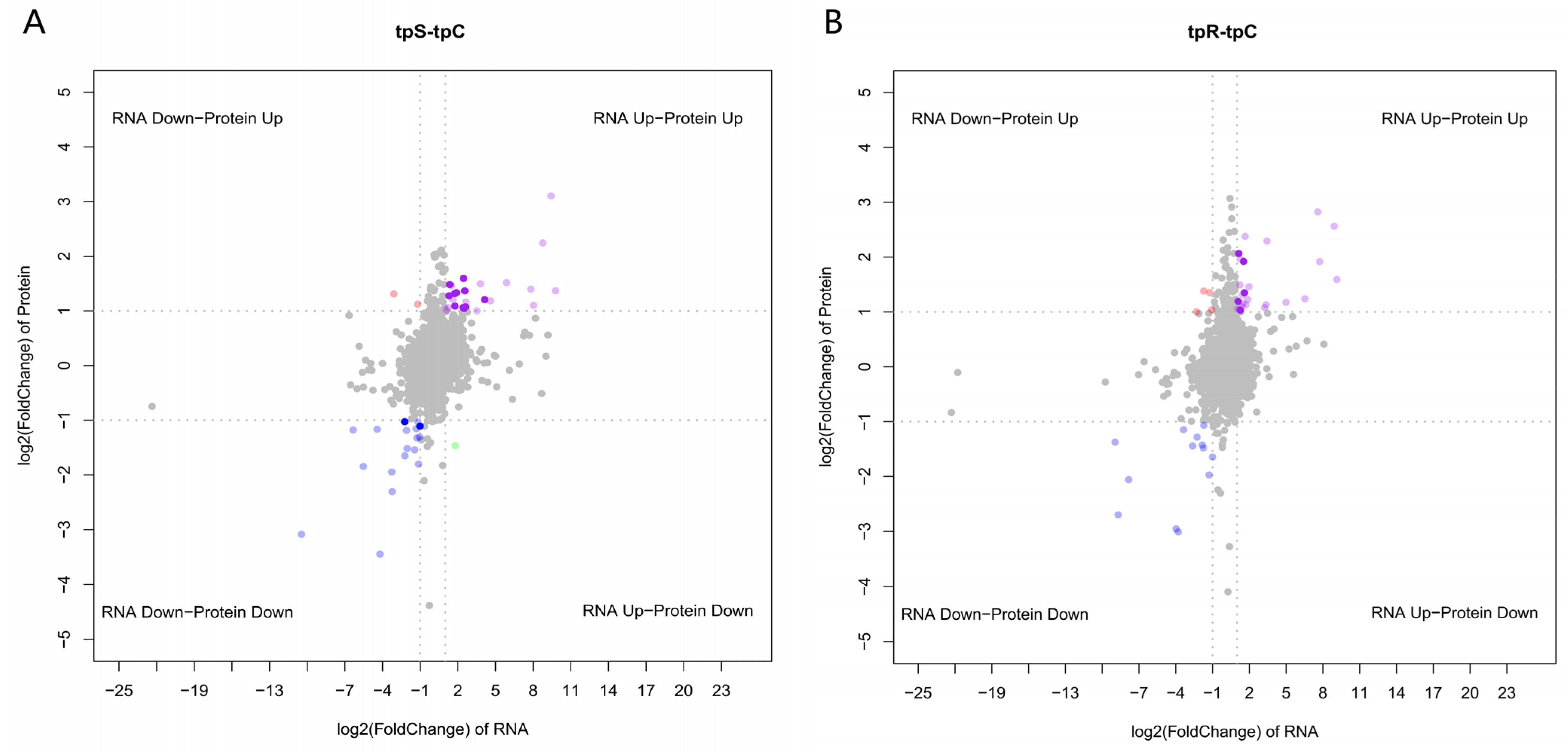
3.5. Correlation Analysis of Transcriptome and Proteome
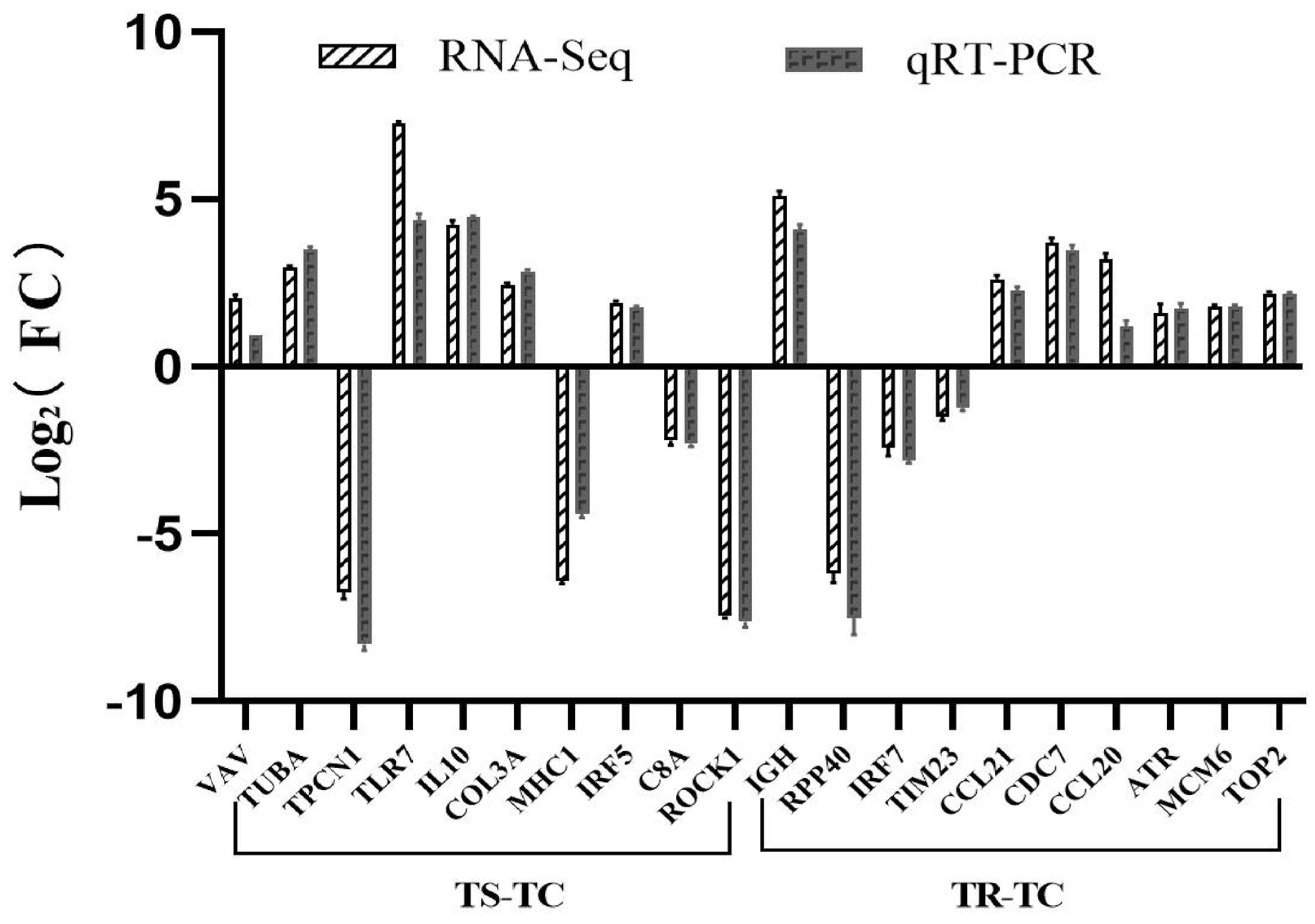
3.6. qRT-PCR Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shen, Y.H.; Ye, Y.Z.; Deng, X.J. Hunan Fauna (Reptiles); Hunan Science and Technology Press: Changsha, China, 2014. [Google Scholar]
- Huang, K.; Bao, J. Progress on Nutritional Composition and Efficacy of Chinese Soft-Shelled Turtle. Acta Agric. Shanghai 2023, 39, 145–152. [Google Scholar]
- Yarahmadi, P.; Miandare, H.K.; Hoseinifar, S.H.; Gheysvandi, N.; Akbarzadeh, A. The effects of stocking density on hemato-immunological and serum biochemical parameters of rainbow trout (Oncorhynchus mykiss). Aquac. Int. 2015, 23, 55–63. [Google Scholar] [CrossRef]
- Natnan, M.E.; Low, C.F.; Chong, C.M.; Bunawan, H.; Baharum, S.N. Integration of omics tools for understanding the fish immune response due to microbial challenge. Front. Mar. Sci. 2021, 8, 668771. [Google Scholar] [CrossRef]
- Zheng, J.F.; Zhang, Y.L.; Zhong, L. Research Progress on Aeromonas hydrophila from Chinese Soft-Shelled Turtle. Fish. Sci. 2016, 35, 191–198. [Google Scholar]
- Luo, F.X.; Mao, P.; Wang, R.H.; Zeng, D.; Xiao, T.Y.; Zhong, L. Pathogenic identification and drug sensitivity analysis of skin fester disease in Trionyx sinensis. J. Aquat. Ecol. 2013, 34, 85–89. [Google Scholar]
- Shen, J.Y.; Pan, X.Y.; Yu, X.P.; Yin, W.L.; Cao, Z.; Wu, Y.L. Pathogen in white abdominal shell disease of soft-shelled turtle (Trionyx sinensis). J. Fish. Sci. China 2007, 5, 815–822. [Google Scholar]
- Jiang, B.J.; Zhang, Z.Q.; Wu, Y.Q.; Yu, J.H.; Huang, Y.; Jian, J.C. Cloning and expression of tox high mobility group box 4 (TOX4) gene in Nile tilapia (Oreochromis niloticus) challenged with Streptococcus agalactiae infection. J. Dalian Ocean. Univ. 2023, 38, 86–94. [Google Scholar]
- Tadiso, T.M.; Krasnov, A.; Skugor, S.; Afanasyev, S.; Hordvik, I.; Nilsen, F. Gene expression analyses of immune responses in Atlantic salmon during early stages of infection by salmon louse (Lepeophtheirus salmonis) reveal bi-phasic responses coinciding with the copepod-chalimus transition. BMC Genom. 2011, 12, 1–17. [Google Scholar] [CrossRef]
- Wang, H.M.; Ding, X.J.; Chen, J.; Song, S.; Tan, B.P.; Zhang, S. Effects of dietary mannan-oligosaccharides on growth performance, serum immune indices, transcriptome and intestinal microflora of Epinephelus fuscoguttatus ♀ × Epinephelus lanceolatus ♂. Acta Nutr. Sin. 2021, 33, 6982–6998. [Google Scholar]
- Hou, S.Q.; Hou, M.N.; Li, J.Y.; Cheng, Y.X. Effects of dietary phospholipid and cholesterol on the hepatopancreas transcriptome of Procambarus clarkii. Genom. Appl. Biol. 2022, 41, 2076–2088. [Google Scholar]
- Zhou, Y.; Li, B.W.; Lin, Q.; Fu, X.Z.; Li, M.; Liang, H.R.; Liu, L.H.; Lu, W.H.; Li, N.Q. Transcriptome analysis of spleen induced by inactivated viral vaccine in marbled sand goby, Oxyeleotris marmoratus. Genom. Appl. Biol. 2019, 38, 4404–4412. [Google Scholar]
- Jaiswal, S.; Rasal, K.D.; Chandra, T.; Prabha, R.; Iquebal, M.A.; Rai, A.; Kumar, D. Proteomics in fish health and aquaculture productivity management: Status and future perspectives. Aquaculture 2023, 566, 739159. [Google Scholar] [CrossRef]
- Cummings, J.H.; Macfarlane, G.T. Collaborative JPEN-Clinical Nutrition Scientific Publications Role of intestinal bacteria in nutrient metabolism. J. Parenter. Enter. Nutr. 1997, 21, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, H.Q.; Niu, H.; Shi, Y.H.; Li, M.Y. Increased liver protein and mRNA expression of natural killer cell-enhancing factor B (NKEF-B) in ayu (Plecoglossus altivelis) after Aeromonas hydrophila infection. Fish Shellfish Immunol. 2009, 26, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Wang, H.C.; Leu, J.H.; Kou, G.H.; Wang, A.H.J.; Lo, C.F. Protein expression profiling of the shrimp cellular response to white spot syndrome virus infection. Dev. Comp. Immunol. 2007, 31, 672–686. [Google Scholar] [CrossRef]
- Jiang, H.; Li, F.H.; Xie, Y.S.; Huang, B.X.; Zhang, J.K.; Zhang, J.Q.; Zhang, C.S.; Li, S.H.; Xiang, J.H. Comparative proteomic profiles of the hepatopancreas in Fenneropenaeus chinensis response to hypoxic stress. Proteomics 2009, 9, 3353–3367. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.W.; Weng, S.P.; Wang, Q.; Shi, X.J.; Dong, C.F.; Lu, Q.X.; Yu, X.Q.; He, J.G. Tiger frog virus can infect zebrafish cells for studying up-or down-regulated genes by proteomics approach. Virus Res. 2009, 144, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Lü, A.; Hu, X.; Wang, Y.; Ming, Q.; Zhu, A.; Shen, L.; Feng, Z. Proteomic analysis of differential protein expression in the skin of zebrafish Danio rerio infected with Aeromonas hydrophila. J. Appl. Ichthyol. 2014, 30, 28–34. [Google Scholar] [CrossRef]
- Peng, X.X. Proteomics and its applications to aquaculture in China: Infection, immunity, and interaction of aquaculture hosts with pathogens. Dev. Comp. Immunol. 2013, 39, 63–71. [Google Scholar] [CrossRef]
- Natnan, M.E.; Mayalvanan, Y.; Jazamuddin, F.M.; Aizat, W.M.; Low, C.-F.; Goh, H.-H.; Azizan, K.A.A.; Bunawan, H.; Baharum, S.N. Omics strategies in current advancements of infectious fish disease management. Biology 2021, 10, 1086. [Google Scholar] [CrossRef]
- Anes, E. Acting on Actin during Bacterial Infection. In Cytoskeleton-Structure, Dynamics, Function and Disease; IntechOpen: London, UK, 2017. [Google Scholar]
- Luo, G.; Zhao, L.; Xu, X.; Qin, Y.; Huang, L.; Su, Y.; Zheng, W.; Yan, Q. Integrated dual RNA-seq and dual iTRAQ of infected tissue reveals the functions of a diguanylate cyclase gene of Pseudomonas plecoglossicida in host-pathogen interactions with Epinephelus coioides. Fish Shellfish Immunol. 2019, 95, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, Y.; Cao, S.; Wang, H.; Zhang, J. Integrated transcriptomic and proteomic analyses of grass carp intestines after vaccination with a double-targeted DNA vaccine of Vibrio mimicus. Fish Shellfish Immunol. 2020, 98, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.D.; Chen, N.; Gao, X.J.; Zhang, Y.Y.; Li, X.X.; Zhang, Y.; Bing, X.W.; Huang, H.Z.; Zhang, X.J. The infection of red seabream iridovirus in mandarin fish (Siniperca chuatsi) and the host immune-related gene expression profiles. Fish Shellfish Immunol. 2018, 74, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Guo, L.; Chen, X.H.; Bian, H.X.; Guo, Y.N.; Jiang, X.J.; He, S.H. Differential proteomic analysis of Mycoplasma synoviae strains of passage 1 and 95. Anim. Husb. Vet. Med. 2021, 53, 52–60. [Google Scholar]
- Laing, K.J.; Dutton, S.; Hansen, J.D. Molecular and biochemical analysis of rainbow trout LCK suggests a conserved mechanism for T-cell signaling in gnathostomes. Mol. Immunol. 2007, 44, 2737–2748. [Google Scholar] [CrossRef]
- Call, M.E.; Wucherpfennig, K.W. Molecular mechanisms for the assembly of the T cell receptor–CD3 complex. Mol. Immunol. 2004, 40, 1295–1305. [Google Scholar] [CrossRef]
- Dave, V.P. Hierarchical role of CD3 chains in thymocyte development. Immunol. Rev. 2009, 232, 22–33. [Google Scholar] [CrossRef]
- Maisey, K.; Toro-Ascuy, D.; Montero, R.; Reyes-López, F.E.; Imarai, M. Identification of CD3ɛ, CD4, CD8β splice variants of Atlantic salmon. Fish Shellfish Immunol. 2011, 31, 815–822. [Google Scholar] [CrossRef]
- Moore, J.C.; Mulligan, T.S.; Yordán, N.T.; Castranova, D.; Pham, V.N.; Tang, Q.; Lobbardi, R.; Anselmo, A.; Liwski, R.S.; Berman, J.N.; et al. T cell immune deficiency in zap70 mutant zebrafish. Mol. Cell. Biol. 2016, 36, 2868–2876. [Google Scholar] [CrossRef]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK–RAS–RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef]
- Terrell, E.M.; Morrison, D.K. Ras-mediated activation of the Raf family kinases. Cold Spring Harb. Perspect. Med. 2019, 9, a033746. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Adjei, A.A. The ras/raf/mapk pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Leulier, F.; Lemaitre, B. Taking an evolutionary approach to Toll-like receptors. Nat. Rev. Genet. 2008, 9, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Beyaert, R. Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol. Cell 2003, 11, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Rao, Y.; Wan, Q.; Liao, Z.; Su, J. Teleost-specific TLR19 localizes to endosome, recognizes dsRNA, recruits TRIF, triggers both IFN and NF-κB pathways, and protects cells from grass carp reovirus infection. J. Immunol. 2018, 200, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, A.; Oshiumi, H.; Tsujita, T.; Mitani, H.; Kasai, H.; Yoshimizu, M.; Matsumoto, M.; Seya, T. Teleost TLR22 recognizes RNA duplex to induce IFN and protect cells from birnaviruses. J. Immunol. 2008, 181, 3474–3485. [Google Scholar] [CrossRef] [PubMed]
- Bi, D.; Wang, Y.; Gao, Y.; Li, X.; Chu, Q.; Cui, J.; Xu, T. Recognition of lipopolysaccharide and activation of NF-κB by cytosolic sensor NOD1 in teleost fish. Front. Immunol. 2018, 9, 1413. [Google Scholar] [CrossRef]
- Pauwels, A.M.; Trost, M.; Beyaert, R.; Hoffmann, E. Patterns, receptors, and signals: Regulation of phagosome maturation. Trends Immunol. 2017, 38, 407–422. [Google Scholar] [CrossRef]
- Weiss, G.E.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef]
- Garin, J.; Diez, R.; Kieffer, S.; Dermine, J.-F.; Duclos, S.; Gagnon, E.; Sadoul, R.; Rondeau, C.; Desjardins, M. The phagosome proteome: Insight into phagosome functions. J. Cell Biol. 2001, 152, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Abbas, Y.M.; Wu, D.; Bueler, S.A.; Robinson, C.V.; Rubinstein, J.L. Structure of V-ATPase from the mammalian brain. Science 2020, 367, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Saito, M.; Tsukahara, A.; Shiokawa, S.; Ueno, K.; Shimamura, H.; Nagano, M.; Toshima, J.Y.; Toshima, J. Functional complementation reveals that 9 of the 13 human V-ATPase subunits can functionally substitute for their yeast orthologs. J. Biol. Chem. 2019, 294, 8273–8285. [Google Scholar] [CrossRef]
- Zhao, J.; Benlekbir, S.; Rubinstein, J.L. Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. Nature 2015, 521, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Wiener, J.J.M.; Wickboldt, A.T.; Wiener, D.K.; Lee-Dutra, A.; Edwards, J.P.; Karlsson, L.; Nguyen, S.; Sun, S.; Jones, T.K.; Grice, C.A. Discovery and SAR of novel pyrazole-based thioethers as cathepsin S inhibitors. Part 2: Modification of P3, P4, and P5 regions. Bioorganic Med. Chem. Lett. 2010, 20, 2375–2378. [Google Scholar] [CrossRef]
- Fonović, U.; Jevnikar, Z.; Kos, J. Cathepsin S generates soluble CX3CL1 (fractalkine) in vascular smooth muscle cells. Biol. Chem. 2013, 394, 1349–1352. [Google Scholar] [CrossRef] [PubMed]
- Thanei, S.; Theron, M.; Silva, A.P.; Reis, B.; Branco, L.; Schirmbeck, L.; Kolb, F.A.; Haap, W.; Schindler, T.; Trendelenburg, M. Cathepsin S inhibition suppresses autoimmune-triggered inflammatory responses in macrophages. Biochem. Pharmacol. 2017, 146, 151–164. [Google Scholar] [CrossRef]
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef]
- Bjorkman, P.J.; Saper, M.A.; Samraoui, B.; Bennett, W.S.; Strominger, J.T.; Wiley, D.C. Structure of the human class I histocompatibility antigen, HLA-A2. Nature 1987, 329, 506–512. [Google Scholar] [CrossRef]
- Cao, X. Medical Immunology, 7th ed.; People’s Medical Publishing House: Beijing, China, 2018; pp. 68–74. [Google Scholar]
- Klein, J. Natural History of the Major Histocompatibility Complex; John Wiley Sons: Hoboken, NJ, USA, 1986. [Google Scholar]
- Li, W.; Jiang, A.; Wu, H. Cloning and Polymorphism Analysis of the MHC Iα Gene in the Yellow Eel. J. Hunan Agric. Univ. Nat. Sci. 2015, 41, 396–400. [Google Scholar]
- Zhang, X.S.; Shen, Y.B.; Xu, X.Y.; Zhang, M.; Bai, Y.L.; Miao, Y.H.; Fang, Y.; Zhang, J.H.; Wang, R.Q.; Li, J.L. Transcriptome analysis and histopathology of black carp (Mylopharyngodon piceus) spleen infected by Aeromonas hydrophila. Fish Shellfish Immunol. 2018, 83, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, C.; Liu, H.; Fu, P. The important role of phagocytosis and interleukins for nile tilapia (Oreochromis niloticus) to defense infection of Aeromonas hydrophila based on transcriptome analysis. Fish Shellfish Immunol. 2019, 92, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, T.; Matsushima, M.; Hashimoto, N.; Imaizumi, K.; Hasegawa, Y. CD40/CD40 ligand interactions in immune responses and pulmonary immunity. Nagoya J. Med. Sci. 2011, 73, 69. [Google Scholar] [PubMed]
- Hassan, S.B.; Sørensen, J.F.; Olsen, B.N.; Pedersen, A.E. Anti-CD40-mediated cancer immunotherapy: An update of recent and ongoing clinical trials. Immunopharmacol. Immunotoxicol. 2014, 36, 96–104. [Google Scholar] [CrossRef]
- Wang, H.M.; Yan, Q.; Yang, T.; Cheng, H.; Du, J.; Yoshioka, K.; Kung, S.K.P.; Ding, G.H. Scaffold protein JLP is critical for CD40 signaling in B lymphocytes. J. Biol. Chem. 2015, 290, 5256–5266. [Google Scholar] [CrossRef]
- Yellin, M.J.; Winikoff, S.; Fortune, S.M.; Baum, D.; Crow, M.K.; Lederman, S.; Chess, L. Ligation of CD40 on fibroblasts induces CD54 (ICAM-1) and CD106 (VCAM-1) up-regulation and IL-6 production and proliferation. J. Leukoc. Biol. 1995, 58, 209–216. [Google Scholar] [CrossRef]
- Schönbeck, U.; Libby, P. The CD40/CD154 receptor/ligand dyad. Cell. Mol. Life Sci. 2001, 58, 4–43. [Google Scholar]
- Wu, Y.; Wang, L.; He, X.; Xu, H.; Zhou, L.; Zhao, F.; Zhang, Y. Expression of CD40 and growth-inhibitory activity of CD40 ligand in colon cancer ex vivo. Cell. Immunol. 2008, 253, 102–109. [Google Scholar] [CrossRef]
- Häkkinen, T.; Karkola, K.; Ylä-Herttuala, S. Macrophages, smooth muscle cells, endothelial cells, and T-cells express CD40 and CD40L in fatty streaks and more advanced human atherosclerotic lesions: Colocalization with epitopes of oxidized low-density lipoprotein, scavenger receptor, and CD16 (FcγRIII). Virchows Arch. 2000, 437, 396–405. [Google Scholar]
- Ma, J.; Chen, T.; Wu, S.; Yang, C.; Bai, M.; Shu, K.; Li, K.; Zhang, G.; Jin, Z.; He, F.; et al. iProX: An integrated proteome resource. Nucleic Acids Res. 2019, 47, D1211–D1217. [Google Scholar] [CrossRef]
- Chen, T.; Ma, J.; Liu, Y.; Chen, Z.G.; Xiao, N.; Lu, Y.T.; Fu, Y.J.; Yang, C.Y.; Li, M.S.; Wu, S.F. iProX in 2021: Connecting proteomics data sharing with big data. Nucleic Acids Res. 2021, 50, D1522–D1527. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Q30 (%) | Mapped Reads (%) |
|---|---|---|---|---|
| TC1 | 110.72 M | 109.32 M | 96.26% | 86,248,137 (78.90%) |
| TC2 | 86.56 M | 85.32 M | 95.80% | 70,666,788 (82.82%) |
| TC3 | 103.01 M | 101.71 M | 96.25% | 85,701,656 (84.26%) |
| TR1 | 100.77 M | 99.40 M | 96.27% | 83,549,364 (84.05%) |
| TR2 | 116.68 M | 115.24 M | 96.27% | 93,791,300 (81.39%) |
| TR3 | 101.50 M | 100.30 M | 96.31% | 82,437,481 (82.19%) |
| TS1 | 113.04 M | 111.54 M | 96.18% | 89,351,292 (80.11%) |
| TS2 | 112.12 M | 110.78 M | 96.34% | 94,055,975 (84.91%) |
| TS3 | 107.27 M | 105.89 M | 96.22% | 86,937,464 (82.10%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ge, L.; Wang, Z.; Hu, Y.; Wang, P.; Qin, Q.; Tian, Y.; Wang, X.; Wen, X.; Zeng, D. Transcriptomic and Proteomic Analyses of the Immune Mechanism in Pathogenetic and Resistant Chinese Soft-Shelled Turtle (Pelodiscus sinensis) Infected with Aeromonas hydrophila. Genes 2024, 15, 1273. https://doi.org/10.3390/genes15101273
Ge L, Wang Z, Hu Y, Wang P, Qin Q, Tian Y, Wang X, Wen X, Zeng D. Transcriptomic and Proteomic Analyses of the Immune Mechanism in Pathogenetic and Resistant Chinese Soft-Shelled Turtle (Pelodiscus sinensis) Infected with Aeromonas hydrophila. Genes. 2024; 15(10):1273. https://doi.org/10.3390/genes15101273
Chicago/Turabian StyleGe, Lingrui, Zi’ao Wang, Yazhou Hu, Pei Wang, Qin Qin, Yu Tian, Xiaoqing Wang, Xingxing Wen, and Dan Zeng. 2024. "Transcriptomic and Proteomic Analyses of the Immune Mechanism in Pathogenetic and Resistant Chinese Soft-Shelled Turtle (Pelodiscus sinensis) Infected with Aeromonas hydrophila" Genes 15, no. 10: 1273. https://doi.org/10.3390/genes15101273