

Combinatorial Anti-Cancer Effect of Polypurine Reverse Hoogsteen Hairpins against KRAS and MYC Targeting in Prostate and Pancreatic Cancer Cell Lines

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
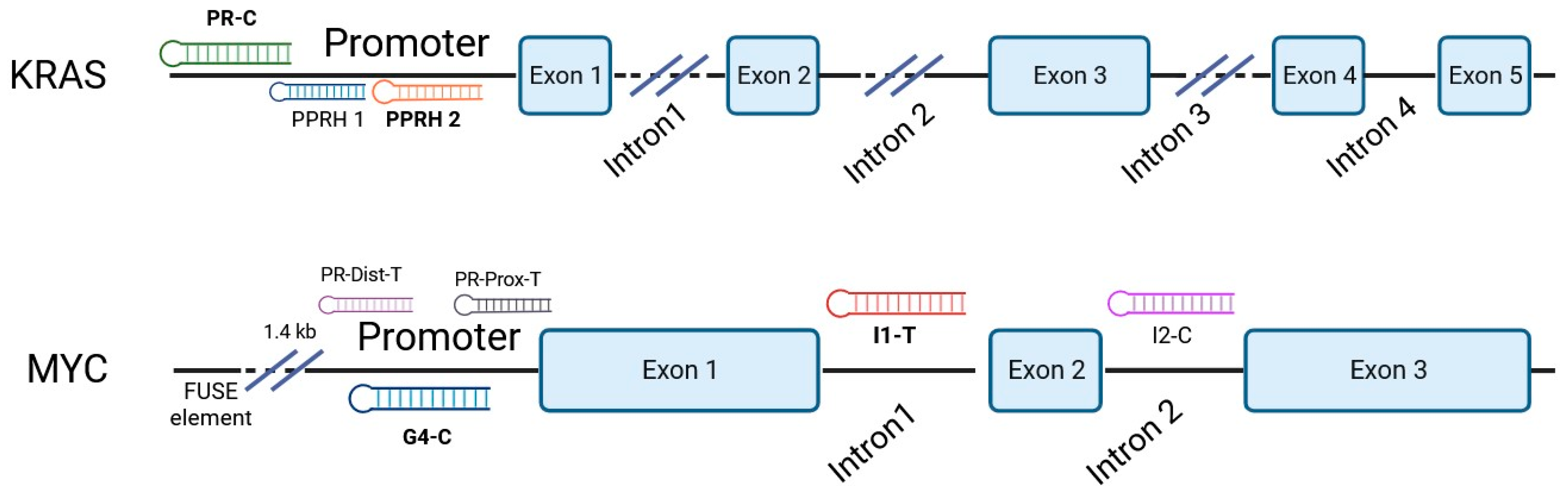
2.1. Polypurine Reverse Hoogsteen Oligonucleotides
2.2. Cell Culture and Cell Transfection
2.3. Cellular Viability with MTT Assay
2.4. Real-Time qPCR
2.5. Western Blot Analyses
2.6. Live Cell Imaging, Confluence, and Cell Cycle Analysis
2.7. Statistical Analyses
3. Results
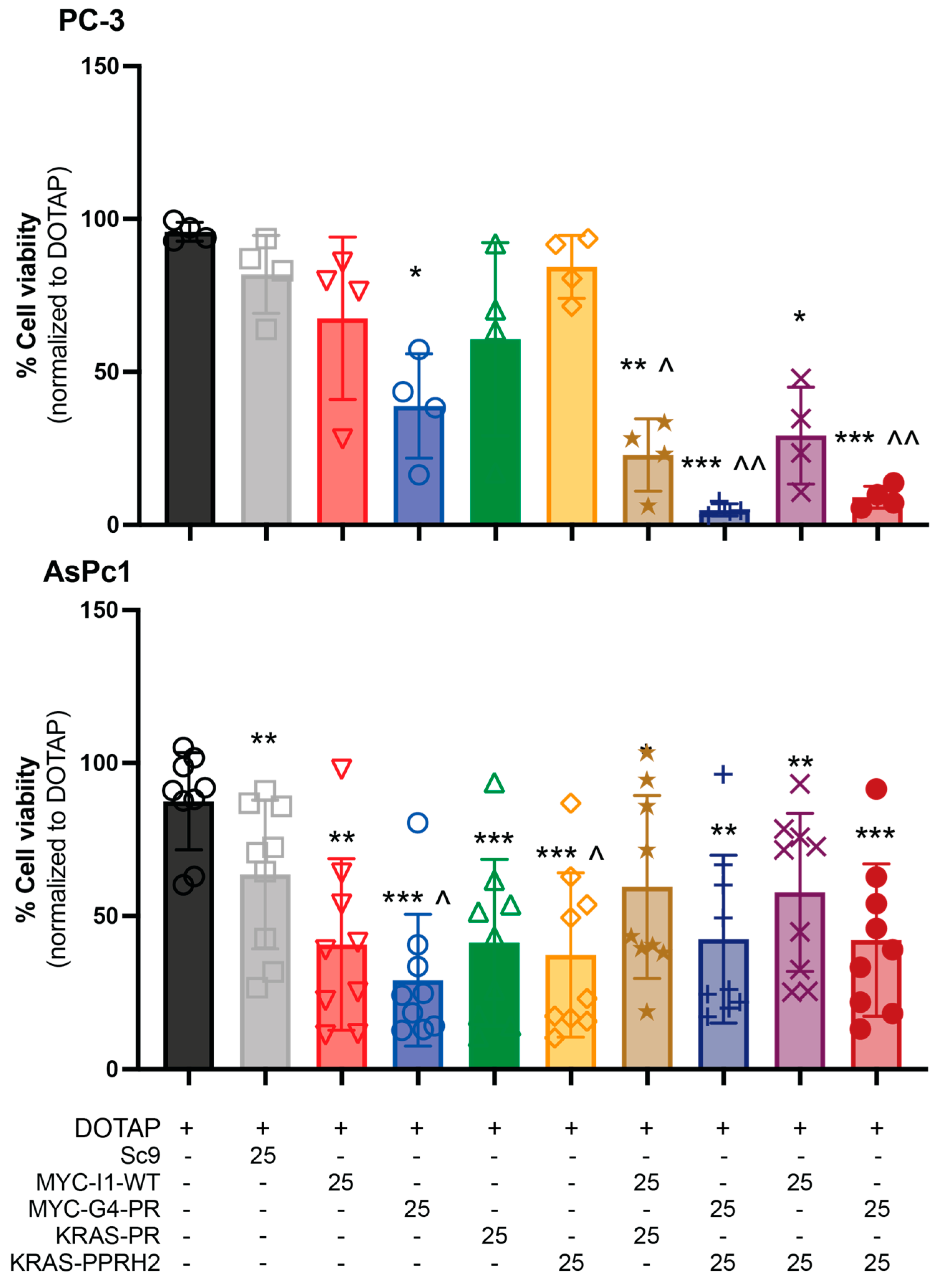
3.1. PPRH Selection and Individual and Combinatorial Effect of PPRHs in PC-3 Cell Viability
3.2. Chemosensitization of Monotherapeutic and Combination PPRHs in PC-3 and AsPc-1 Cells
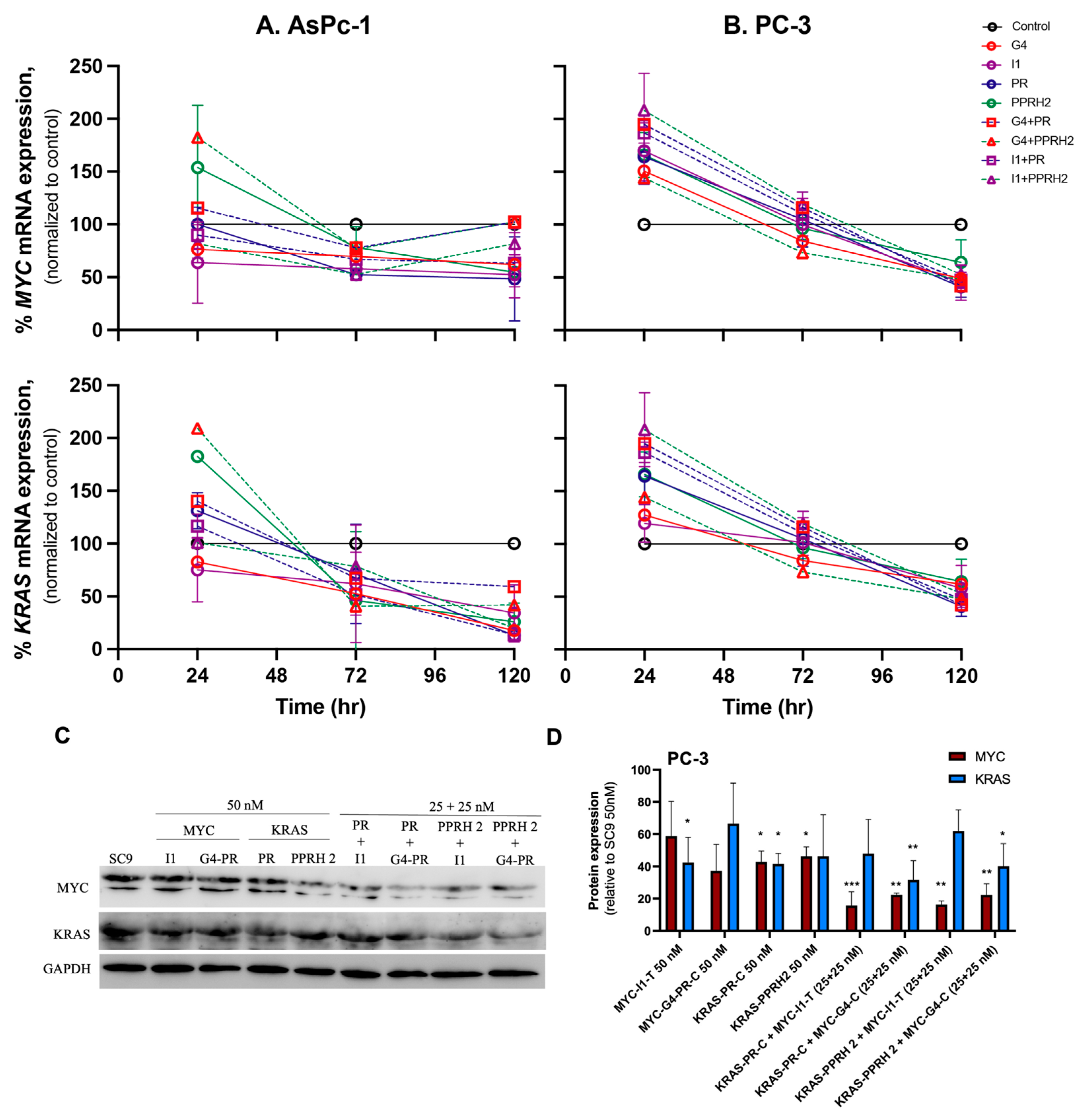
3.3. Individual and Combinatorial Effects of PPRHs on KRAS and MYC mRNA and Protein Levels
3.4. PPRH Cell Penetrance, Effect on Growth and the Cell Cycle
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adjei, A.A. Blocking Oncogenic Ras Signaling for Cancer Therapy. J. Natl. Cancer Inst. 2001, 93, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Colicelli, J. Human RAS Superfamily Proteins and Related GTPases. Sci. STKE 2004, 2004, RE13. [Google Scholar] [CrossRef] [PubMed]
- Pantsar, T. The Current Understanding of KRAS Protein Structure and Dynamics. Comput. Struct. Biotechnol. J. 2020, 18, 189. [Google Scholar] [CrossRef] [PubMed]
- Friday, B.B.; Adjei, A.A. K-Ras as a Target for Cancer Therapy. Biochim. Biophys. Acta 2005, 1756, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS Mutation: From Undruggable to Druggable in Cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Zhu, G.; Pei, L.; Xia, H.; Tang, Q.; Bi, F. Role of Oncogenic KRAS in the Prognosis, Diagnosis and Treatment of Colorectal Cancer. Mol. Cancer 2021, 20, 143. [Google Scholar] [CrossRef]
- Nissley, D.V.; McCormick, F. RAS at 40: Update from the RAS Initiative. Cancer Discov. 2022, 12, 895–898. [Google Scholar] [CrossRef]
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C Mutated Metastatic NSCLC. Clin. Cancer Res. 2022, 28, 1482–1486. [Google Scholar] [CrossRef]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘Undruggable’ Cancer Targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Neumann, J.; Zeindl-Eberhart, E.; Kirchner, T.; Jung, A. Frequency and Type of KRAS Mutations in Routine Diagnostic Analysis of Metastatic Colorectal Cancer. Pathol. Res. Pract. 2009, 205, 858–862. [Google Scholar] [CrossRef]
- Salem, M.E.; El-Refai, S.M.; Sha, W.; Puccini, A.; Grothey, A.; George, T.J.; Hwang, J.J.; O’Neil, B.; Barrett, A.S.; Kadakia, K.C.; et al. Landscape of KRASG12C, Associated Genomic Alterations, and Interrelation With Immuno-Oncology Biomarkers in KRAS-Mutated Cancers. JCO Precis. Oncol. 2022, 6, e2100245. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A Gene Expression Signature Associated with “K-Ras Addiction” Reveals Regulators of EMT and Tumor Cell Survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, S.; Kaushik, G.; Nimmakayala, R.; Rachagani, S.; Ponnusamy, M.P.; Batra, S.K. MicroRNA regulation of K-Ras in pancreatic cancer and opportunities for therapeutic intervention. Semin. Cancer Biol. 2019, 54, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ou, Y.; Wu, K.; Chen, Y.; Sun, W. MiR-143 Inhibits the Metastasis of Pancreatic Cancer and an Associated Signaling Pathway. Tumour Biol. 2012, 33, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Guan, X.; Zhang, X.; Luan, X.; Song, Z.; Cheng, X.; Zhang, W.; Qin, J.J. Targeting KRAS Mutant Cancers: From Druggable Therapy to Drug Resistance. Mol. Cancer 2022, 21, 159. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; O’Grady, S.; Tang, M.; Crown, J. MYC as a Target for Cancer Treatment. Cancer Treat. Rev. 2021, 94, 102154. [Google Scholar] [CrossRef]
- Beaulieu, M.E.; Castillo, F.; Soucek, L. Structural and Biophysical Insights into the Function of the Intrinsically Disordered Myc Oncoprotein. Cells 2020, 9, 1038. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 Years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Armelin, H.A.; Armelin, M.C.S.; Kelly, K.; Stewart, T.; Leder, P.; Cochran, B.H.; Stiles, C.D. Functional Role for C-Myc in Mitogenic Response to Platelet-Derived Growth Factor. Nature 1984, 310, 655–660. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Shen, L.; O’Shea, J.M.; Kaadige, M.R.; Cunha, S.; Wilde, B.R.; Cohen, A.L.; Welm, A.L.; Ayer, D.E. Metabolic Reprogramming in Triple-Negative Breast Cancer through Myc Suppression of TXNIP. Proc. Natl. Acad. Sci. USA 2015, 112, 5425–5430. [Google Scholar] [CrossRef] [PubMed]
- Herrick, D.J.; Ross, J. The Half-Life of c-Myc MRNA in Growing and Serum-Stimulated Cells: Influence of the Coding and 3′ Untranslated Regions and Role of Ribosome Translocation. Mol. Cell. Biol. 1994, 14, 2119–2128. [Google Scholar] [CrossRef] [PubMed]
- Levens, D.L. How the C-Myc Promoter Works and Why It Sometimes Does Not. J. Natl. Cancer Inst. Monogr. 2008, 2008, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Llombart, V.; Mansour, M.R. Therapeutic Targeting of “Undruggable” MYC. EBioMedicine 2022, 75, 103756. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The Landscape of Somatic Copy-Number Alteration across Human Cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Mossafa, H.; Damotte, D.; Jenabian, A.; Delarue, R.; Vincenneau, A.; Amouroux, I.; Jeandel, R.; Khoury, E.; Martelli, J.M.; Samson, T.; et al. Non-Hodgkin’s Lymphomas with Burkitt-like Cells Are Associated with c-Myc Amplification and Poor Prognosis. Leuk. Lymphoma 2006, 47, 1885–1893. [Google Scholar] [CrossRef]
- Vita, M.; Henriksson, M. The Myc Oncoprotein as a Therapeutic Target for Human Cancer. Semin. Cancer Biol. 2006, 16, 318–330. [Google Scholar] [CrossRef]
- McCormick, F. KRAS as a Therapeutic Target. Clin. Cancer Res. 2015, 21, 1797–1801. [Google Scholar] [CrossRef]
- Whitfield, J.R.; Beaulieu, M.E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef]
- Waters, A.M.; Khatib, T.O.; Papke, B.; Goodwin, C.M.; Hobbs, G.A.; Diehl, J.N.; Yang, R.; Edwards, A.C.; Walsh, K.H.; Sulahian, R.; et al. Targeting P130Cas- and Microtubule-Dependent MYC Regulation Sensitizes Pancreatic Cancer to ERK MAPK Inhibition. Cell Rep. 2021, 35, 109291. [Google Scholar] [CrossRef]
- Hashimoto, A.; Handa, H.; Hata, S.; Tsutaho, A.; Yoshida, T.; Hirano, S.; Hashimoto, S.; Sabe, H. Inhibition of Mutant KRAS-Driven Overexpression of ARF6 and MYC by an EIF4A Inhibitor Drug Improves the Effects of Anti-PD-1 Immunotherapy for Pancreatic Cancer. Cell Commun. Signal. 2021, 19, 54. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Blake, D.R.; Gilbert, T.S.K.; Ng, S.; Hostetter, G.; Azam, S.H.; Ozkan-Dagliyan, I.; Gautam, P.; Bryant, K.L.; Pearce, K.H.; et al. KRAS Suppression-Induced Degradation of MYC Is Antagonized by a MEK5-ERK5 Compensatory Mechanism. Cancer Cell 2018, 34, 807–822.e7. [Google Scholar] [CrossRef] [PubMed]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The Regulation and Functions of DNA and RNA G-Quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA Secondary Structures: Stability and Function of G-Quadruplex Structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef]
- Huppert, J.L.; Balasubramanian, S. G-Quadruplexes in Promoters throughout the Human Genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef]
- Linke, R.; Limmer, M.; Juranek, S.A.; Heine, A.; Paeschke, K. The Relevance of G-Quadruplexes for DNA Repair. Int. J. Mol. Sci. 2021, 22, 12599. [Google Scholar] [CrossRef]
- Sarkies, P.; Reams, C.; Simpson, L.J.; Sale, J.E. Epigenetic Instability Due to Defective Replication of Structured DNA. Mol. Cell 2010, 40, 703–713. [Google Scholar] [CrossRef]
- Brooks, T.A.; Hurley, L.H. Targeting MYC Expression through G-Quadruplexes. Genes Cancer 2010, 1, 641–649. [Google Scholar] [CrossRef]
- Dexheimer, T.S.; Sun, D.; Hurley, L.H. Deconvoluting the Structural and Drug-Recognition Complexity of the G-Quadruplex-Forming Region Upstream of the Bcl-2 P1 Promoter. J. Am. Chem. Soc. 2006, 128, 5404–5415. [Google Scholar] [CrossRef]
- Cogoi, S.; Xodo, L.E. G-Quadruplex Formation within the Promoter of the KRAS Proto-Oncogene and Its Effect on Transcription. Nucleic Acids Res. 2006, 34, 2536–2549. [Google Scholar] [CrossRef] [PubMed]
- Coma, S.; Noé, V.; Eritja, R.; Ciudad, C.J. Strand Displacement of Double- Stranded DNA by Triplex-Forming Antiparallel Purine-Hairpins. Oligonucleotides 2005, 15, 269–283. [Google Scholar] [CrossRef] [PubMed]
- de Almagro, M.C.; Coma, S.; Noé, V.; Ciudad, C.J. Polypurine Hairpins Directed against the Template Strand of DNA Knock Down the Expression of Mammalian Genes. J. Biol. Chem. 2009, 284, 11579–11589. [Google Scholar] [CrossRef] [PubMed]
- Noé, V.; Aubets, E.; Félix, A.J.; Ciudad, C.J. Nucleic Acids Therapeutics Using PolyPurine Reverse Hoogsteen Hairpins. Biochem. Pharmacol. 2021, 189, 114371. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, L.; Villalobos, X.; Solé, A.; Lliberós, C.; Ciudad, C.J.; Noé, V. Improved Design of PPRHs for Gene Silencing. Mol. Pharm. 2015, 12, 867–877. [Google Scholar] [CrossRef]
- De Almagro, M.C.; Mencia, N.; Noé, V.; Ciudad, C.J. Coding Polypurine Hairpins Cause Target-Induced Cell Death in Breast Cancer Cells. Hum. Gene Ther. 2011, 22, 451–463. [Google Scholar] [CrossRef]
- Villalobos, X.; Rodríguez, L.; Solé, A.; Lliberós, C.; Mencia, N.; Ciudad, C.J.; Noé, V. Effect of Polypurine Reverse Hoogsteen Hairpins on Relevant Cancer Target Genes in Different Human Cell Lines. Nucleic Acid Ther. 2015, 25, 198–208. [Google Scholar] [CrossRef]
- López-Aguilar, E.; Fernández-Nogueira, P.; Fuster, G.; Carbó, N.; Ciudad, C.J.; Noé, V. In Vitro and In Vivo Effects of the Combination of Polypurine Reverse Hoogsteen Hairpins against HER-2 and Trastuzumab in Breast Cancer Cells. Int. J. Mol. Sci. 2023, 24, 7073. [Google Scholar] [CrossRef]
- Aubets, E.; Félix, A.J.; Garavís, M.; Reyes, L.; Aviñó, A.; Eritja, R.; Ciudad, C.J.; Noé, V. Detection of a G-Quadruplex as a Regulatory Element in Thymidylate Synthase for Gene Silencing Using Polypurine Reverse Hoogsteen Hairpins. Int. J. Mol. Sci. 2020, 21, 5028. [Google Scholar] [CrossRef]
- Psaras, A.M.; Valiuska, S.; Noé, V.; Ciudad, C.J.; Brooks, T.A. Targeting KRAS Regulation with PolyPurine Reverse Hoogsteen Oligonucleotides. Int. J. Mol. Sci. 2022, 23, 2097. [Google Scholar] [CrossRef]
- Valiuska, S.; Psaras, A.M.; Noé, V.; Brooks, T.A.; Ciudad, C.J. Targeting MYC Regulation with Polypurine Reverse Hoogsteen Oligonucleotides. Int. J. Mol. Sci. 2022, 24, 378. [Google Scholar] [CrossRef] [PubMed]
- Papadimitropoulou, A.; Makri, M.; Zoidis, G. MYC the Oncogene from Hell: Novel Opportunities for Cancer Therapy. Eur. J. Med. Chem. 2024, 267, 116194. [Google Scholar] [CrossRef] [PubMed]
- Garralda, E.; Beaulieu, M.-E.; Moreno, V.; Casacuberta-Serra, S.; Martínez-Martín, S.; Foradada, L.; Alonso, G.; Massó-Vallés, D.; López-Estévez, S.; Jauset, T.; et al. Nature Medicine MYC Targeting by OMO-103 in Solid Tumors: A Phase 1 Trial. Nat. Med. 2024, 30, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, pii: baw100. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; Von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and Repression by Oncogenic MYC Shape Tumour-Specific Gene Expression Profiles. Nature 2014, 511, 483–487. [Google Scholar] [CrossRef]
- Psaras, A.M.; Chang, K.T.; Hao, T.; Brooks, T.A. Targeted Downregulation of MYC through G-Quadruplex Stabilization by DNAi. Molecules 2021, 26, 5542. [Google Scholar] [CrossRef]
- Phillips, P.C. Epistasis--the Essential Role of Gene Interactions in the Structure and Evolution of Genetic Systems. Nat. Rev. Genet. 2008, 9, 855–867. [Google Scholar] [CrossRef]
- Eilers, M.; Eisenman, R.N. Myc’s Broad Reach. Genes Dev. 2008, 22, 2755–2766. [Google Scholar] [CrossRef]
- Ischenko, I.; Zhi, J.; Hayman, M.J.; Petrenko, O. KRAS-Dependent Suppression of MYC Enhances the Sensitivity of Cancer Cells to Cytotoxic Agents. Oncotarget 2017, 8, 17995–18009. [Google Scholar] [CrossRef]
- Lee, T.; Yao, G.; Nevins, J.; You, L. Sensing and Integration of Erk and PI3K Signals by Myc. PLoS Comput. Biol. 2008, 4, e1000013. [Google Scholar] [CrossRef] [PubMed]
- Shortt, J.; Johnstone, R.W. Oncogenes in Cell Survival and Cell Death. Cold Spring Harb. Perspect. Biol. 2012, 4, a009829. [Google Scholar] [CrossRef]
- Chang, W.H.; Liu, Y.; Hammes, E.A.; Bryant, K.L.; Cerione, R.A.; Antonyak, M.A. Oncogenic RAS Promotes MYC Protein Stability by Upregulating the Expression of the Inhibitor of Apoptosis Protein Family Member Survivin. J. Biol. Chem. 2023, 299, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Donati, G.; Amati, B. MYC and Therapy Resistance in Cancer: Risks and Opportunities. Mol. Oncol. 2022, 16, 3828–3854. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | PPRH Name (Location) | Length | Sequence (5′-3′) PPRH + Target |
|---|---|---|---|
| KRAS | HpKRAS-PR-C (Promoter) | 52 | 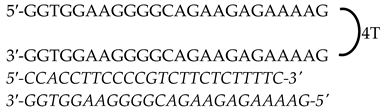 |
| HpKRAS-PR-EF-C (PPRH 1) (Promoter) | 42 | 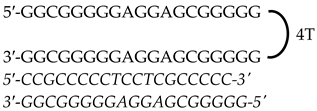 | |
| HpKRAS-PR-BC-C (PPRH 2) (Promoter) | 46 | 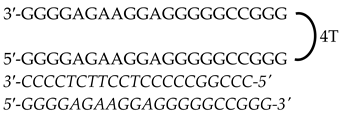 | |
| MYC | HpMYC-G4-PR-C (Promoter) | 67 |  |
| HpMYC-PR-Distal-T (Promoter) | 50 |  | |
| HpMYC-PR-Prox-T (Promoter) | 50 | 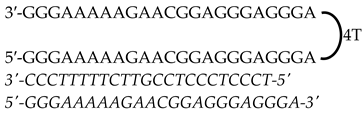 | |
| HpMYC-I1-T (Intron 1) | 72 | 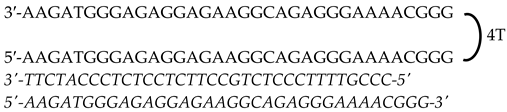 | |
| HpMYC-I2-C (Intron 2) | 52 |  |
| Dose MYC PPRHs (nM) | Dose KRAS PPRHs (nM) | CI Media | Description | ||
|---|---|---|---|---|---|
| I1-T | G4-C | PR-C | PPRH2 | ||
| 25 | - | 25 | - | 0.88 | Slight synergism |
| 25 | - | - | 25 | 0.65 | Synergism |
| - | 25 | 25 | - | 0.57 | Synergism |
| - | 25 | - | 25 | 0.41 | Synergism |
| AsPc-1: TXL IC50 (μM) | PC-3: CIS IC50 (μM) | |
|---|---|---|
| DOTAP | 0.68 | 1.734 |
| G4-C | 0.1627 | 1.862 |
| I1-T | 0.2016 | 1.527 |
| PR-C | 0.3135 | 2.041 |
| PPRH2 | 0.2426 | 1.856 |
| G4-C+ PR-C | 0.2211 | 1.733 |
| G4-C + PPRH2 | 0.2739 | 0.7278 |
| I1-T + PR-C | 0.3938 | 1.422 |
| I1-T + PPRH2 | 0.2622 | 1.541 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valiuska, S.; Elder, K.K.; McKay, S.J.; Ciudad, C.J.; Noé, V.; Brooks, T.A. Combinatorial Anti-Cancer Effect of Polypurine Reverse Hoogsteen Hairpins against KRAS and MYC Targeting in Prostate and Pancreatic Cancer Cell Lines. Genes 2024, 15, 1332. https://doi.org/10.3390/genes15101332
Valiuska S, Elder KK, McKay SJ, Ciudad CJ, Noé V, Brooks TA. Combinatorial Anti-Cancer Effect of Polypurine Reverse Hoogsteen Hairpins against KRAS and MYC Targeting in Prostate and Pancreatic Cancer Cell Lines. Genes. 2024; 15(10):1332. https://doi.org/10.3390/genes15101332
Chicago/Turabian StyleValiuska, Simonas, Kayla K. Elder, Steven J. McKay, Carlos J. Ciudad, Véronique Noé, and Tracy A. Brooks. 2024. "Combinatorial Anti-Cancer Effect of Polypurine Reverse Hoogsteen Hairpins against KRAS and MYC Targeting in Prostate and Pancreatic Cancer Cell Lines" Genes 15, no. 10: 1332. https://doi.org/10.3390/genes15101332
APA StyleValiuska, S., Elder, K. K., McKay, S. J., Ciudad, C. J., Noé, V., & Brooks, T. A. (2024). Combinatorial Anti-Cancer Effect of Polypurine Reverse Hoogsteen Hairpins against KRAS and MYC Targeting in Prostate and Pancreatic Cancer Cell Lines. Genes, 15(10), 1332. https://doi.org/10.3390/genes15101332