
Identification of Novel QTL for Mercury Accumulation in Maize Using an Enlarged SNP Panel

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Field Trials
2.2. Measurement of Hg Content
2.3. Genotype and GWAS
- (1)
- Multiple statistical models, namely 5PCs + K, Q, K, and Q + K, were employed. The 5PCs + K model controls both the top five principal components (PCs) and the kinship matrix, as used in the study by Zhao et al. [27]. The Q model controls only the population structure, the K model solely controls the kinship matrix, and the Q + K model controls both the population structure and the kinship matrix. These analyses utilized the 0.55 M genotypic datasets, and the optimal statistical model was chosen based on quantile–quantile (QQ) plots.
- (2)
- GWAS results under 0.55 M were compared between 5PCs + K and the optimal statistical model to assess whether changing the model could enhance the statistical power of GWAS.
- (3)
- Under the optimal statistical model, GWAS was conducted using an enlarged genotypic dataset (1.25 M). The results from GWAS with the 0.55 M and 1.25 M datasets were compared to determine if increasing the marker density improved the statistical power of the analysis.
- (4)
- Finally, the GWAS results of the optimal statistical model under the enlarged 1.25 M genotypic dataset were used for subsequent analysis. The effective marker number (En) of the two genotypic datasets was calculated using GEC V1.0 software. The result indicated that En was 490,548 for the 1.25 M dataset and 250,345 for the 0.55 M dataset. In addition, the software suggested a significance threshold of p ≤ (1/En), which was used for the association analysis.
2.4. Candidate Gene Identification
2.5. Analysis of Expression Level Association of Candidate Genes
2.6. Haplotype Analyses of Candidate Genes
3. Results
3.1. Model Comparison and Selection
3.2. Boosting GWAS Power through Increased Marker Density
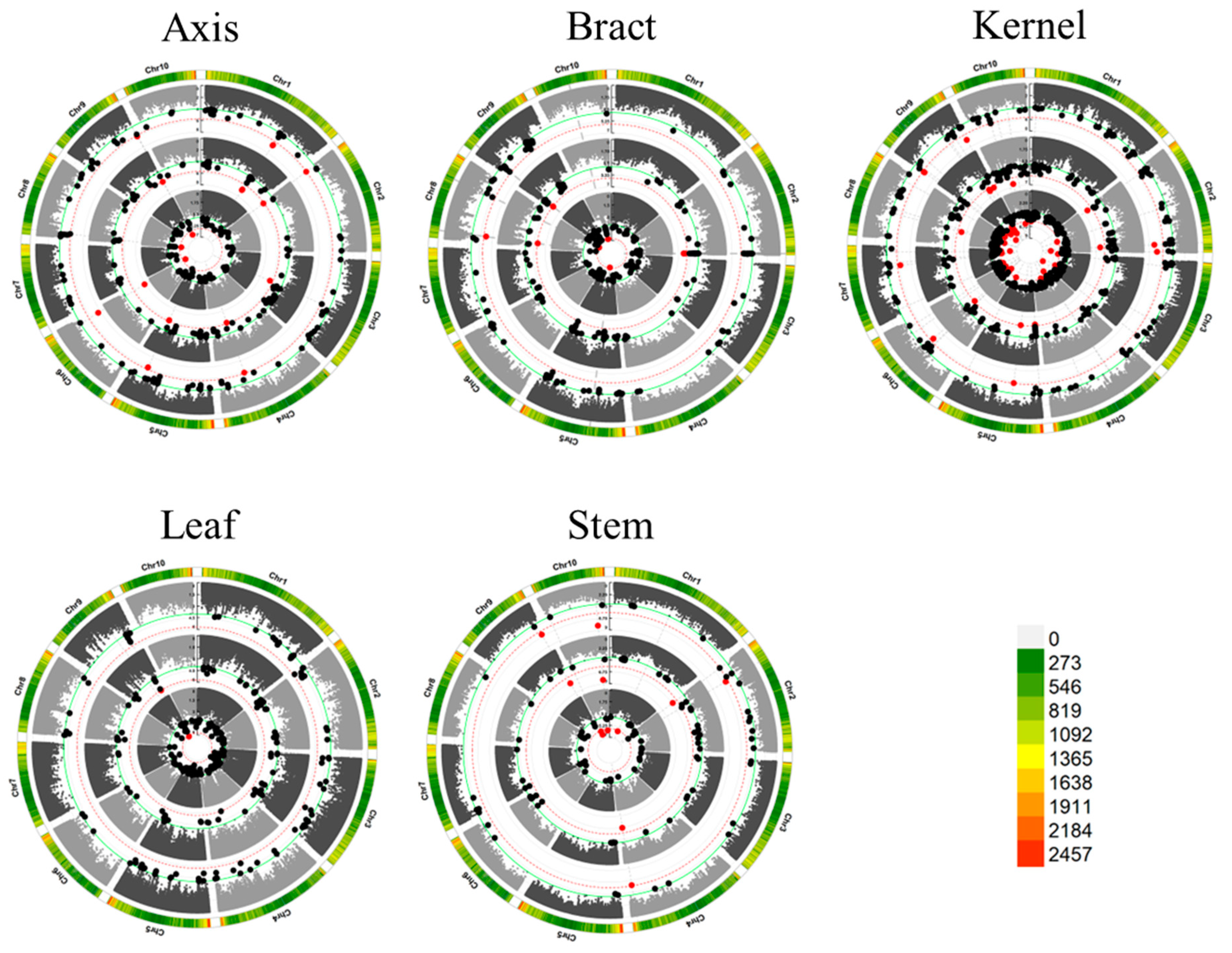
3.3. Significant Loci and Tissue-Specific Variability
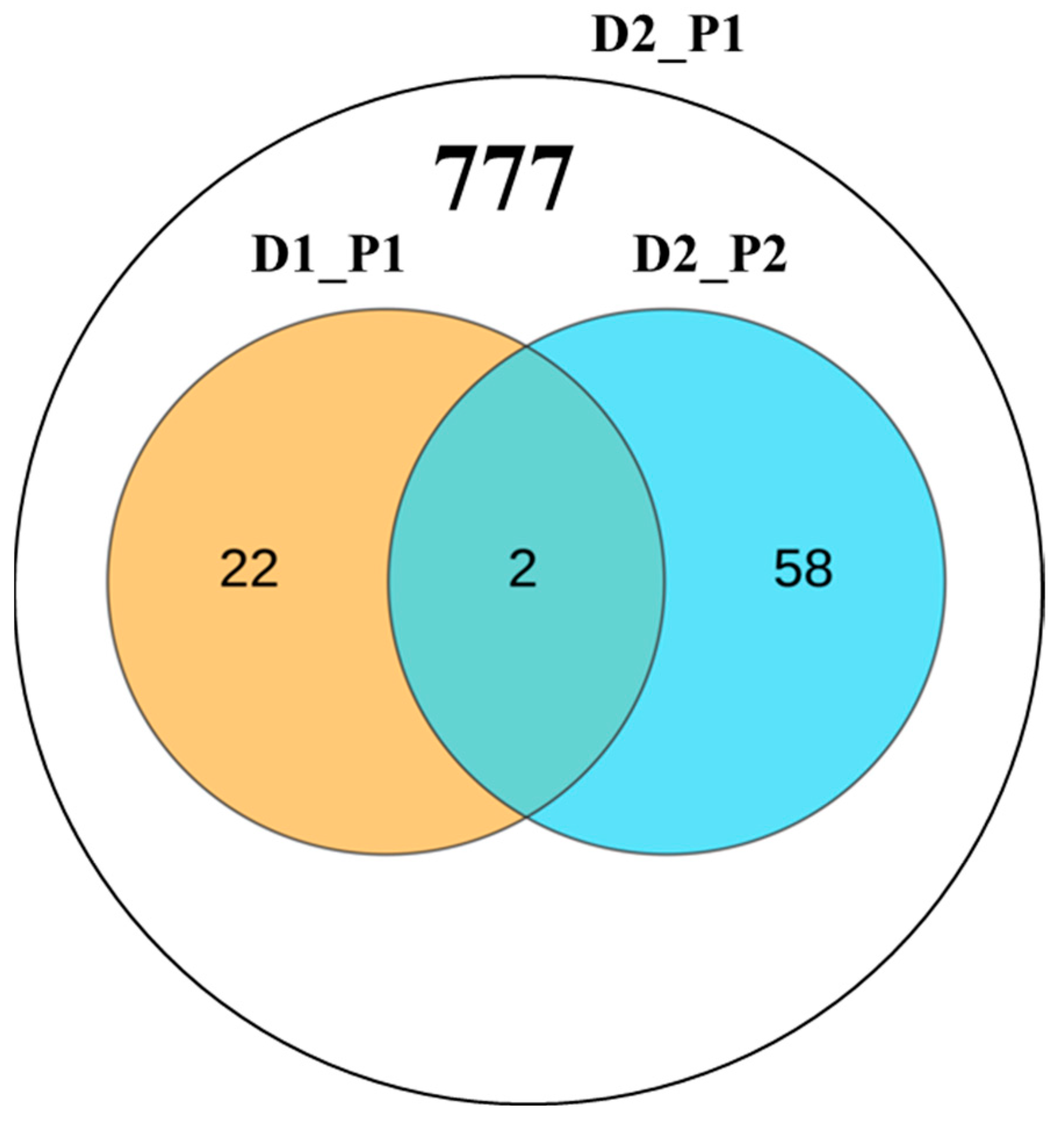
3.4. Colocalized Loci Identified across Various Tissues and Environments
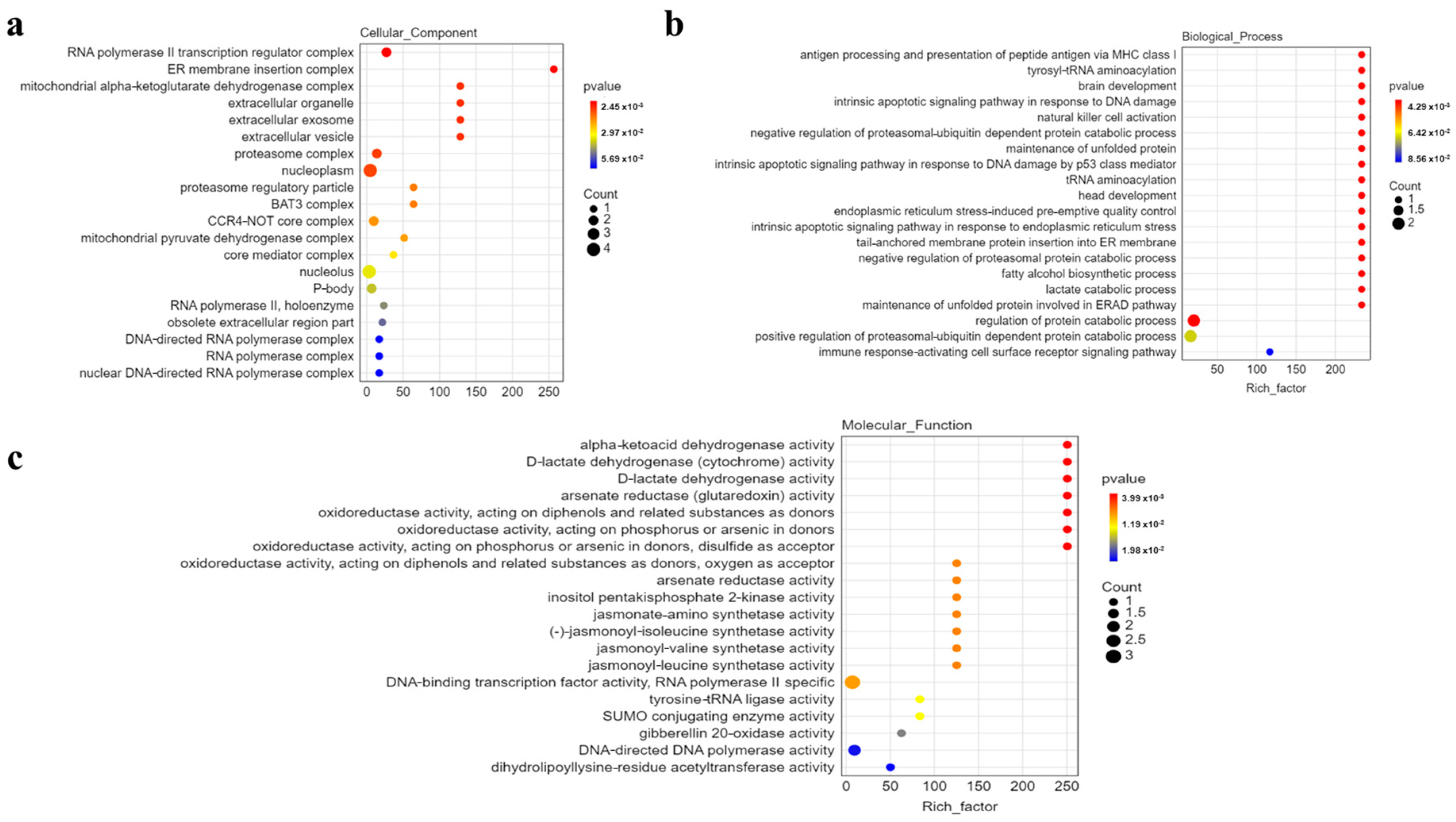
3.5. Functional Analysis of Identified Genes
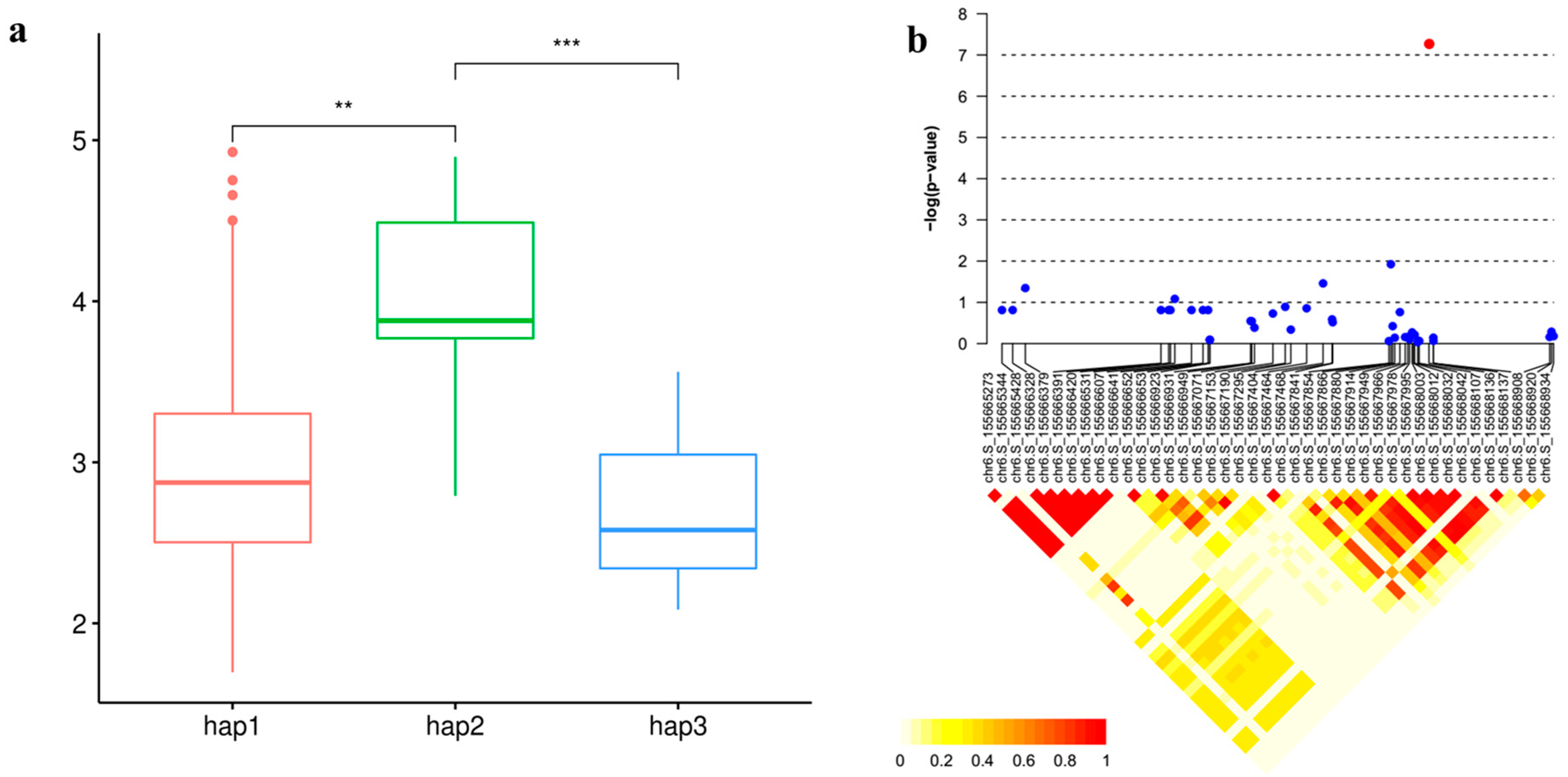
3.6. Candidate Gene Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahman, Z.; Singh, V.P. The relative impact of toxic heavy metals (THMs) (arsenic (As); cadmium (Cd); chromium (Cr)(VI); mercury (Hg); and lead (Pb)) on the total environment: An overview. Environ. Monit. Assess. 2019, 191, 419. [Google Scholar] [CrossRef]
- Shao, R.; Zhang, J.; Shi, W.; Wang, Y.; Tang, Y.; Liu, Z.; Sun, W.; Wang, H.; Guo, J.; Meng, Y.; et al. Mercury stress tolerance in wheat and maize is achieved by lignin accumulation controlled by nitric oxide. Environ. Pollut. 2022, 307, 119488. [Google Scholar] [CrossRef]
- Boerleider, R.Z.; Roeleveld, N.; Scheepers, P. Human biological monitoring of mercury for exposure assessment. AIMS Environ. Sci. 2017, 4, 251–276. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Z.; Gao, Y.; Zhang, X. Planular-vertical distribution and pollution characteristics of cropland soil Hg and the estimated soil-air exchange fluxes of gaseous Hg over croplands in northern China. Environ. Res. 2021, 195, 110810. [Google Scholar] [CrossRef]
- Zhang, L.; Wong, M.H. Environmental mercury contamination in China: Sources and impacts. Environ. Int. 2017, 33, 108–121. [Google Scholar] [CrossRef]
- Sun, T.; Wang, Z.; Zhang, X.; Niu, Z.; Chen, J. Influences of high-level atmospheric gaseous elemental mercury on methylmercury accumulation in maize (Zea mays L.). Environ. Pollut. 2020, 265 Pt B, 114890. [Google Scholar] [CrossRef]
- Liu, S.; Wang, X.; Guo, G.; Yan, Z. Status and environmental management of soil mercury pollution in China: A review. J. Environ. Manag. 2021, 277, 111442. [Google Scholar] [CrossRef]
- Rocha, J.; Aschner, M.; Dorea, J.G.; Ceccatelli, S.; Farina, M.; Silveira, L. Mercury toxicity. J. Biomed. Biotechnol. 2012, 2012, 831890. [Google Scholar] [CrossRef]
- Wang, C.; Wang, T.; Mu, P.; Li, Z.; Yang, L. Quantitative Trait Loci for Mercury Tolerance in Rice Seedlings. Rice Sci. 2013, 20, 238–242. [Google Scholar] [CrossRef]
- Fu, Z.; Li, W.; Zhang, Q.; Wang, L.; Zhang, X.; Song, G.; Fu, Z.; Ding, D.; Liu, Z.; Tang, J. Quantitative trait loci for mercury accumulation in maize (Zea mays L.) identified using a RIL population. PLoS ONE 2014, 9, e107243. [Google Scholar] [CrossRef]
- Hu, T.; Liu, Y.; Zhu, S.; Qin, J.; Li, W.; Zhou, N. Overexpression of OsLea14-A improves the tolerance of rice and increases Hg accumulation under diverse stresses. Environ. Sci. Pollut. Res. Int. 2019, 26, 10537–10551. [Google Scholar] [CrossRef]
- Sun, L.; Ma, Y.; Wang, H.; Huang, W.; Wang, X.; Han, L.; Sun, W.; Han, E.; Wang, B. Overexpression of PtABCC1 contributes to mercury tolerance and accumulation in Arabidopsis and poplar. Biochem. Biophys. Res. Commun. 2018, 497, 997–1002. [Google Scholar] [CrossRef]
- Xu, S.; Sun, B.; Wang, R.; He, J.; Xia, B.; Xue, Y.; Wang, R. Overexpression of a bacterial mercury transporter MerT in Arabidopsis enhances mercury tolerance. Biochem. Biophys. Res. Commun. 2017, 490, 528–534. [Google Scholar] [CrossRef]
- Yu, J.; Buckler, E.S. Genetic association mapping and genome organization of maize. Curr. Opin. Biotechnol. 2006, 17, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Liu, H.; Wu, L.; Warburton, M.; Yan, J. Genome-wide Association Studies in Maize: Praise and Stargaze. Mol. Plant 2017, 10, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Peng, Z.; Yang, X.; Wang, W.; Fu, J.; Wang, J.; Han, Y.; Chai, Y.; Guo, T.; Yang, N.; et al. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 2013, 45, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Beló, A.; Zheng, P.; Luck, S.; Shen, B.; Meyer, D.J.; Li, B.; Tingey, S.; Rafalski, A. Whole genome scan detects an allelic variant of fad2 associated with increased oleic acid levels in maize. Mol. Genet. Genom. 2008, 279, 1–10. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, H.; Fu, Z.; Chen, H.; Lin, Y.; Yan, P.; Li, W.; Xie, H.; Guo, Z.; Zhang, X.; et al. Genetic-based dissection of arsenic accumulation in maize using a genome-wide association analysis method. Plant Biotechnol. J. 2018, 16, 1085–1093. [Google Scholar] [CrossRef]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, Y.; Wei, X.; Li, C.; Wang, A.; Zhao, Q.; Li, W.; Guo, Y.; Deng, L.; Zhu, C.; et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 2011, 44, 32–39. [Google Scholar] [CrossRef]
- Wang, S.B.; Feng, J.Y.; Ren, W.L.; Huang, B.; Zhou, L.; Wen, Y.J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef]
- Wang, Q.; Tian, F.; Pan, Y.; Buckler, E.S.; Zhang, Z. A SUPER powerful method for genome wide association study. PLoS ONE 2014, 9, e107684. [Google Scholar] [CrossRef]
- Li, X.; Zhu, C.; Yeh, C.T.; Wu, W.; Takacs, E.M.; Petsch, K.A.; Tian, F.; Bai, G.; Buckler, E.S.; Muehlbauer, G.J.; et al. Genic and nongenic contributions to natural variation of quantitative traits in maize. Genome Res. 2012, 22, 2436–2444. [Google Scholar] [CrossRef]
- Yang, N.; Lu, Y.; Yang, X.; Huang, J.; Zhou, Y.; Ali, F.; Wen, W.; Liu, J.; Li, J.; Yan, J. Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet. 2014, 10, e1004573. [Google Scholar] [CrossRef]
- Tian, F.; Bradbury, P.; Brown, P.; Hung, H.; Sun, Q.; Flint-Garcia, S.; Rocheford, T.R.; McMullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 2011, 43, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Tung, C.W.; Eizenga, G.C.; Wright, M.H.; Ali, M.L.; Price, A.H.; Norton, G.J.; Islam, M.R.; Reynolds, A.; Mezey, J.; et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2011, 2, 467. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Fu, Z.; Lin, Y.; Chen, H.; Liu, K.; Xing, X.; Liu, Z.; Li, W.; Tang, J. Genome-wide association analysis identifies loci governing mercury accumulation in maize. Sci. Rep. 2017, 7, 247. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, S.; Fan, Y.; Liu, N.; Zhan, W.; Liu, H.; Xiao, Y.; Li, K.; Pan, Q.; Li, W.; et al. Beyond pathways: Genetic dissection of tocopherol content in maize kernels by combining linkage and association analyses. Plant Biotechnol. J. 2018, 16, 1464–1475. [Google Scholar] [CrossRef]
- Liu, H.; Luo, X.; Niu, L.; Xiao, Y.; Chen, L.; Liu, J.; Wang, X.; Jin, M.; Li, W.; Zhang, Q.; et al. Distant eQTLs and Non-coding Sequences Play Critical Roles in Regulating Gene Expression and Quantitative Trait Variation in Maize. Mol. Plant 2017, 10, 414–426. [Google Scholar] [CrossRef]
- Li, Q.; Yang, X.; Xu, S.; Cai, Y.; Zhang, D.; Han, Y.; Li, L.; Zhang, Z.; Gao, S.; Li, J.; et al. Genome-wide association studies identified three independent polymorphisms associated with alpha-tocopherol content in maize kernels. PLoS ONE 2012, 7, e36807. [Google Scholar]
- Fu, J.; Cheng, Y.; Linghu, J.; Yang, X.; Kang, L.; Zhang, Z.; Zhang, J.; He, C.; Du, X.; Peng, Z.; et al. RNA sequencing reveals the complex regulatory network in the maize kernel. Nat. Commun. 2013, 4, 2832. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.; Glaubitz, J.; Sun, Q.; Poland, J.; Kawamoto, K.; Buckler, E.; Mitchell, S. A robust; simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Deng, M.; Li, D.; Luo, J.; Xiao, Y.; Liu, H.; Pan, Q.; Zhang, X.; Jin, M.; Zhao, M.; Yan, J. The genetic architecture of amino acids dissection by association and linkage analysis in maize. Plant Biotechnol. J. 2017, 15, 1250–1263. [Google Scholar] [CrossRef]
- Liu, H.; Wang, X.; Warburton, M.L.; Wen, W.; Jin, M.; Deng, M.; Liu, J.; Tong, H.; Pan, Q.; Yang, X.; et al. Genomic; Transcriptomic; and Phenomic Variation Reveals the Complex Adaptation of Modern Maize Breeding. Mol. Plant 2015, 8, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, B.; Yang, Z.; Liu, Y.; Yang, S.; Shi, Y.; Jiang, C.; Qin, F. Manipulating ZmEXPA4 expression ameliorates the drought-induced prolonged anthesis and silking interval in maize. Plant Cell 2021, 33, 2058–2071. [Google Scholar] [CrossRef]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef]
- Khorchid, A.; Ikura, M. Bacterial histidine kinase as signal sensor and transducer. Int. J. Biochem. Cell Biol. 2006, 38, 307–312. [Google Scholar] [CrossRef]
- Yu, X.; Meng, X.; Xu, M.; Zhang, X.; Zhang, Y.; Ding, G.; Huang, S.; Zhang, A.; Jia, Z. Celastrol ameliorates cisplatin nephrotoxicity by inhibiting NF-kappaB and improving mitochondrial function. EBioMedicine 2018, 36, 266–280. [Google Scholar] [CrossRef]
- Stratton, A.; Ericksen, M.; Harris, T.V.; Symmonds, N.; Silverstein, T.P. Mercury(II) binds to both of chymotrypsin’s histidines, causing inhibition followed by irreversible denaturation/aggregation. Protein Sci. 2017, 26, 292–305. [Google Scholar] [CrossRef]
- Cao, Y.; Ma, C.; Chen, H.; Chen, G.; White, J.C.; Xing, B. Copper stress in flooded soil: Impact on enzyme activities; microbial community composition and diversity in the rhizosphere of Salix integra. Sci. Total Environ. 2020, 704, 135350. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.N.; Liu, B.Y.; Ma, Y.C.; Yang, H.L.; Liu, G.Q. Analysis of reference genes stability and histidine kinase expression under cold stress in Cordyceps militaris. PLoS ONE 2020, 15, e0236898. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Verslues, P. Stress physiology functions of the Arabidopsis histidine kinase cytokinin receptors. Physiol. Plant 2015, 154, 369–380. [Google Scholar] [CrossRef]
- Abbehausen, C. Zinc finger domains as therapeutic targets for metal-based compounds—An update. Metallomics 2019, 11, 15–28. [Google Scholar] [CrossRef]
- Banerjee, M.; Ferragut Cardoso, A.P.; Lykoudi, A.; Wilkey, D.W.; Pan, J.; Watson, W.H.; Garbett, N.C.; Rai, S.N.; Merchant, M.L.; States, J.C. Arsenite Exposure Displaces Zinc from ZRANB2 Leading to Altered Splicing. Chem. Res. Toxicol. 2020, 33, 1403–1417. [Google Scholar] [CrossRef]
- Razmiafshari, M.; Kao, J.; d’Avignon, A.; Zawia, N.H. NMR identification of heavy metal-binding sites in a synthetic zinc finger peptide: Toxicological implications for the interactions of xenobiotic metals with zinc finger proteins. Toxicol. Appl. Pharmacol. 2001, 172, 1–10. [Google Scholar] [CrossRef]
- Sivo, V.; D’Abrosca, G.; Baglivo, I.; Iacovino, R.; Pedone, P.V.; Fattorusso, R.; Russo, L.; Malgieri, G.; Isernia, C. Ni(II); Hg(II); and Pb(II) Coordination in the Prokaryotic Zinc-Finger Ros87. Inorg. Chem. 2019, 58, 1067–1080. [Google Scholar] [CrossRef]
- Zhang, X.; Warburton, M.L.; Setter, T.; Liu, H.; Xue, Y.; Yang, N.; Yan, J.; Xiao, Y. Genome-wide association studies of drought-related metabolic changes in maize using an enlarged SNP panel. Theor. Appl. Genet. 2016, 129, 1449–1463. [Google Scholar] [CrossRef]
- Tibbs Cortes, L.; Zhang, Z.; Yu, J. Status and prospects of genome-wide association studies in plants. Plant Genome 2021, 14, e20077. [Google Scholar] [CrossRef]
- Liao, X.; Sun, J.; Li, Q.; Ding, W.; Zhao, B.; Wang, B.; Zhou, S.; Wang, H. ZmSIZ1a and ZmSIZ1b play an indispensable role in resistance against Fusarium ear rot in maize. Mol. Plant Pathol. 2023, 24, 711–724. [Google Scholar] [CrossRef]
- Kumari, N.; Jagadevan, S. Genetic identification of arsenate reductase and arsenite oxidase in redox transformations carried out by arsenic metabolising prokaryotes—A comprehensive review. Chemosphere 2016, 163, 400–412. [Google Scholar] [CrossRef]
- An, B.; Lan, J.; Deng, X.; Chen, S.; Ouyang, C.; Shi, H.; Yang, J.; Li, Y. Silencing of D-Lactate Dehydrogenase Impedes Glyoxalase System and Leads to Methylglyoxal Accumulation and Growth Inhibition in Rice. Front. Plant Sci. 2017, 8, 2071. [Google Scholar] [CrossRef]
- Sun, Y.; Xu, W.; Wu, L.; Wang, R.; He, Z.; Ma, M. An Arabidopsis mutant of inositol pentakisphosphate 2-kinase AtIPK1 displays reduced arsenate tolerance. Plant Cell Environ. 2016, 39, 416–426. [Google Scholar] [CrossRef]
- Maruyama, K.; Yorifuji, T.; Tsuda, T.; Sekikawa, T.; Nakadaira, H.; Saito, H. Methyl mercury exposure at Niigata; Japan: Results of neurological examinations of 103 adults. J. Biomed. Biotechnol. 2012, 2012, 635075. [Google Scholar] [CrossRef]
- Huang, C.; Liu, S.; Lin-Shiau, S. Neurotoxicological effects of cinnabar (a Chinese mineral medicine; HgS) in mice. Toxicol. Appl. Pharmacol. 2007, 224, 192–201. [Google Scholar] [CrossRef]
- Canabady-Rochelle, L.L.; Harscoat-Schiavo, C.; Kessler, V.; Aymes, A.; Fournier, F.; Girardet, J.M. Determination of reducing power and metal chelating ability of antioxidant peptides: Revisited methods. Food Chem. 2015, 183, 129–135. [Google Scholar] [CrossRef]
- Torres-Fuentes, C.; Alaiz, M.; Vioque, J. Iron-chelating activity of chickpea protein hydrolysate peptides. Food Chem. 2012, 134, 1585–1588. [Google Scholar] [CrossRef]
- Chang, J.; Zhou, Y.; Wang, Q.; Aschner, M.; Lu, R. Plant components can reduce methylmercury toxication: A mini-review. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 129290. [Google Scholar] [CrossRef]
- Li, Y.; Takada, M.; Mizuno, N. Premotor neurons projecting simultaneously to two orofacial motor nuclei by sending their branched axons. A study with a fluorescent retrograde double-labeling technique in the rat. Neurosci. Lett. 1993, 152, 29–32. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID a | Chr. | Position b | Trait | Location | SNP | p Value c | R2 d | Candidate Gene e | Annotation | Expressed or Not f | Verified by Expression GWAS g |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 4 | 167403585 | Axis | BLUP | chr4.S_167403585 | 1.54 × 10−6 | 0.18 | GRMZM2G125943 | histidine kinase | express | Ture |
| GRMZM2G125991 | endoglucanase 7-like | express | Ture | ||||||||
| GRMZM2G029859 | pentatricopeptide repeat-containing protein At3g16610 | express | Ture | ||||||||
| 2 | 4 | 167403585 | Axis | XX | chr4.S_167403585 | 1.33 × 10−6 | 0.18 | GRMZM2G125943 | histidine kinase | express | Ture |
| GRMZM2G125991 | endoglucanase 7-like | express | Ture | ||||||||
| GRMZM2G029859 | pentatricopeptide repeat-containing protein At3g16610 | express | Ture | ||||||||
| 3 | 5 | 2356674 | Kernel | XX | chr5.S_2356674 | 1.31 × 10−6 | 0.10 | GRMZM2G002825 | actin-depolymerizing factor 3 | express | Ture |
| GRMZM2G002805 | zinc finger protein ZAT5 | express | Ture | ||||||||
| GRMZM2G002815 | NA | express | Ture | ||||||||
| GRMZM2G144188 | dof zinc finger protein DOF2.4-like | not | |||||||||
| GRMZM2G144172 | dof zinc finger protein DOF2.4-like | express | Ture | ||||||||
| GRMZM2G003068 | NA | express | Ture | ||||||||
| GRMZM2G003108 | CRAL/TRIO domain containing protein | express | Ture | ||||||||
| 4 | 6 | 155668107 | Axis | BLUP | chr6.S_155668107 | 2.82 × 10−8 | 0.13 | GRMZM2G566873 | NA | not | |
| GRMZM2G140805 | NA | not | |||||||||
| GRMZM2G440949 | dr1-associated corepressor | express | Ture | ||||||||
| GRMZM2G440968 | cystatin 3 | express | Ture | ||||||||
| GRMZM2G140817 | putative cytochrome P450 superfamily protein | express | Ture | ||||||||
| 5 | 6 | 155668107 | Axis | CG | chr6.S_155668107 | 6.77 × 10−7 | 0.10 | GRMZM2G566873 | NA | not | |
| GRMZM2G140805 | NA | not | |||||||||
| GRMZM2G440949 | dr1-associated corepressor | express | Ture | ||||||||
| GRMZM2G440968 | cystatin 3 | express | Ture | ||||||||
| GRMZM2G140817 | putative cytochrome P450 superfamily protein | express | Ture | ||||||||
| 6 | 6 | 155668107 | Axis | XX | chr6.S_155668107 | 1.46 × 10−8 | 0.13 | GRMZM2G566873 | NA | not | |
| GRMZM2G140805 | NA | not | |||||||||
| GRMZM2G440949 | dr1-associated corepressor | express | Ture | ||||||||
| GRMZM2G440968 | cystatin 3 | express | Ture | ||||||||
| GRMZM2G140817 | putative cytochrome P450 superfamily protein | express | Ture | ||||||||
| 7 | 10 | 127359876 | Stem | BLUP | chr10.S_127359876 | 8.01 × 10−9 | 0.23 | GRMZM2G005633 | Endochitinase B | express | Ture |
| GRMZM2G006428 | NA | express | Ture | ||||||||
| GRMZM2G006216 | S-adenosyl-L-methionine-dependent methyltransferase superfamily protein | express | Ture | ||||||||
| GRMZM2G005939 | NA | express | Ture | ||||||||
| 8 | 10 | 127359876 | Stem | CG | chr10.S_127359876 | 1.11 × 10−6 | 0.17 | GRMZM2G005633 | Endochitinase B | express | Ture |
| GRMZM2G006428 | NA | express | Ture | ||||||||
| GRMZM2G006216 | S-adenosyl-L-methionine-dependent methyltransferase superfamily protein | express | Ture | ||||||||
| GRMZM2G005939 | NA | express | Ture | ||||||||
| 9 | 10 | 127359876 | Stem | XX | chr10.S_127359876 | 5.81 × 10−9 | 0.24 | GRMZM2G005633 | Endochitinase B | express | Ture |
| GRMZM2G006428 | NA | express | Ture | ||||||||
| GRMZM2G006216 | S-adenosyl-L-methionine-dependent methyltransferase superfamily protein | express | Ture | ||||||||
| GRMZM2G005939 | NA | express | Ture |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Li, J.; Zhang, J.; Sun, Y.; Ju, X.; Li, W.; Duan, H.; Xue, Z.; Sun, L.; Hussain Sahito, J.; et al. Identification of Novel QTL for Mercury Accumulation in Maize Using an Enlarged SNP Panel. Genes 2024, 15, 257. https://doi.org/10.3390/genes15020257
Gao J, Li J, Zhang J, Sun Y, Ju X, Li W, Duan H, Xue Z, Sun L, Hussain Sahito J, et al. Identification of Novel QTL for Mercury Accumulation in Maize Using an Enlarged SNP Panel. Genes. 2024; 15(2):257. https://doi.org/10.3390/genes15020257
Chicago/Turabian StyleGao, Jionghao, Jianxin Li, Jihong Zhang, Yan Sun, Xiaolong Ju, Wenlong Li, Haiyang Duan, Zhengjie Xue, Li Sun, Javed Hussain Sahito, and et al. 2024. "Identification of Novel QTL for Mercury Accumulation in Maize Using an Enlarged SNP Panel" Genes 15, no. 2: 257. https://doi.org/10.3390/genes15020257
APA StyleGao, J., Li, J., Zhang, J., Sun, Y., Ju, X., Li, W., Duan, H., Xue, Z., Sun, L., Hussain Sahito, J., Fu, Z., Zhang, X., & Tang, J. (2024). Identification of Novel QTL for Mercury Accumulation in Maize Using an Enlarged SNP Panel. Genes, 15(2), 257. https://doi.org/10.3390/genes15020257