
Transcriptome Responses to Different Salinity Conditions in Litoditis marina, Revealed by Long-Read Sequencing


Abstract
1. Introduction
2. Material and Methods
2.1. Worms
2.2. Sample Collection and ONT lrRNA Sequencing Library Preparation
2.3. Transcriptome Construction from ONT Reads
2.4. Gene and Transcript Abundance Estimation
2.5. Differential Expression Analysis
2.6. Alternative Splicing Analysis
2.7. Alternative Polyadenylation Analysis
2.8. Analysis of 3′ UTR
2.9. Gene Set and Pathway Enrichment Analyses
3. Result
3.1. Overview of ONT lrRNA Sequencing in Different Salinity Conditions
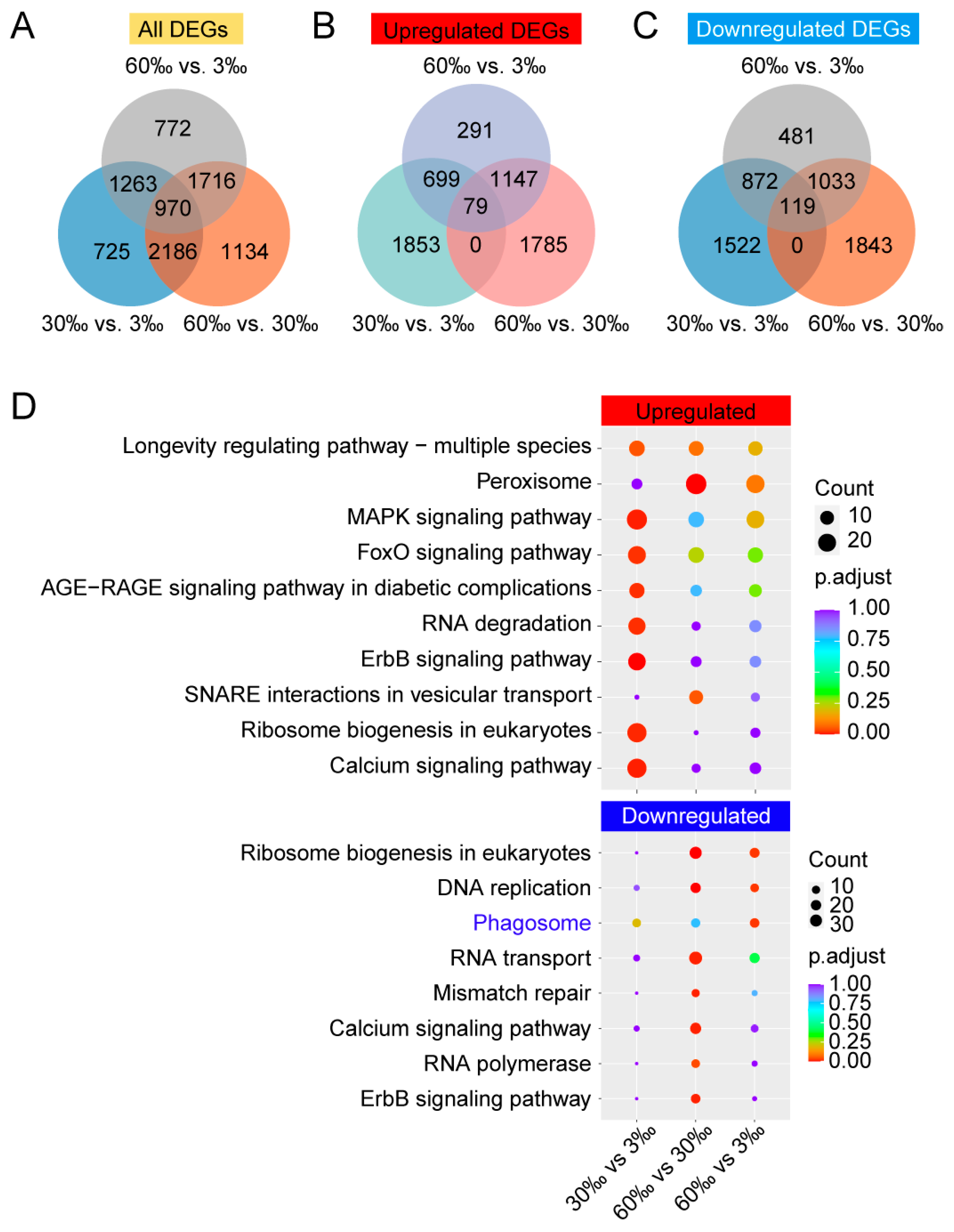
3.2. Differentially Expressed Genes (DEGs) Analysis
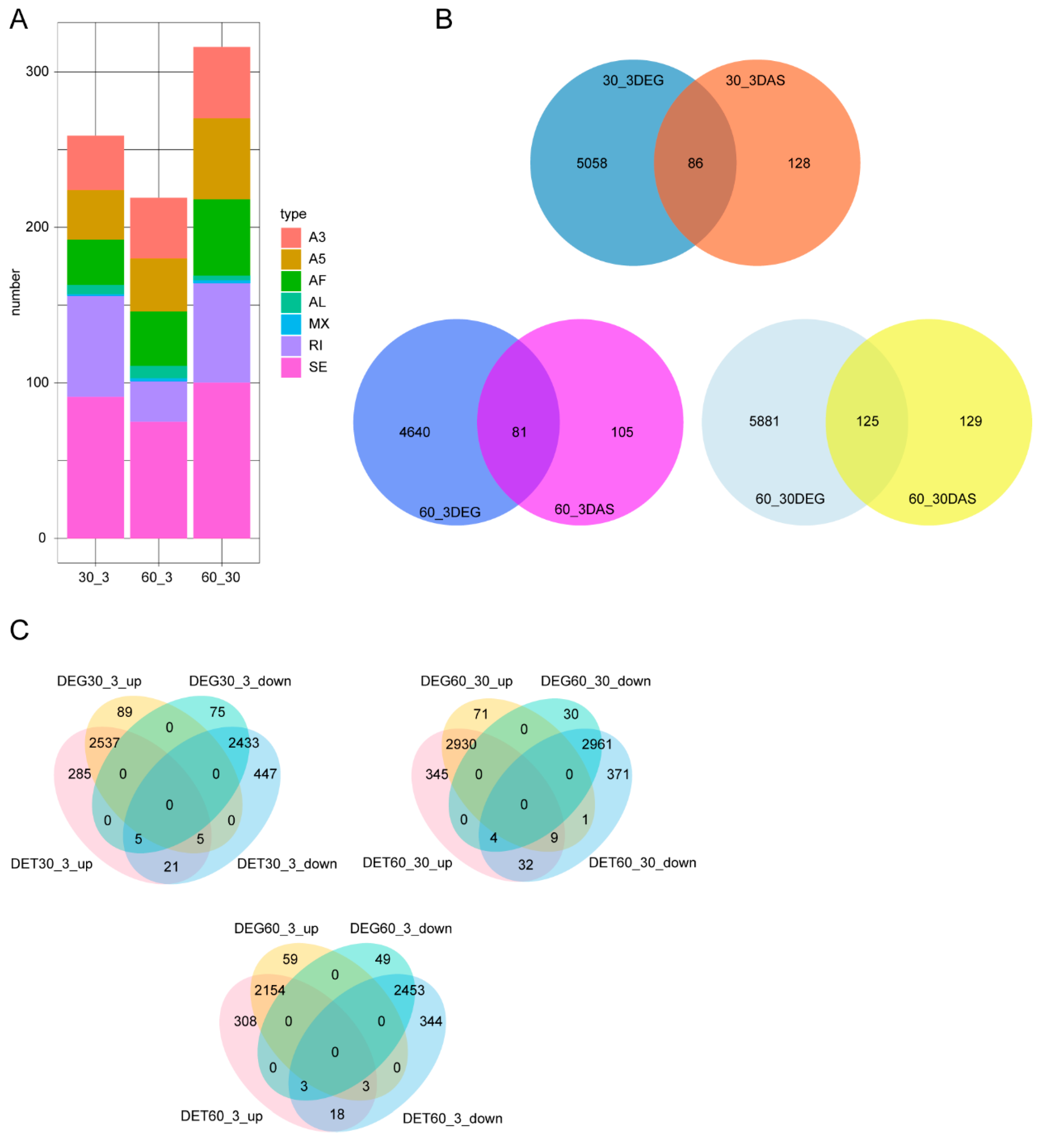
3.3. Analysis of Differential Alternative Splicing (DAS) and Differentially Expressed Genes (DEGs)
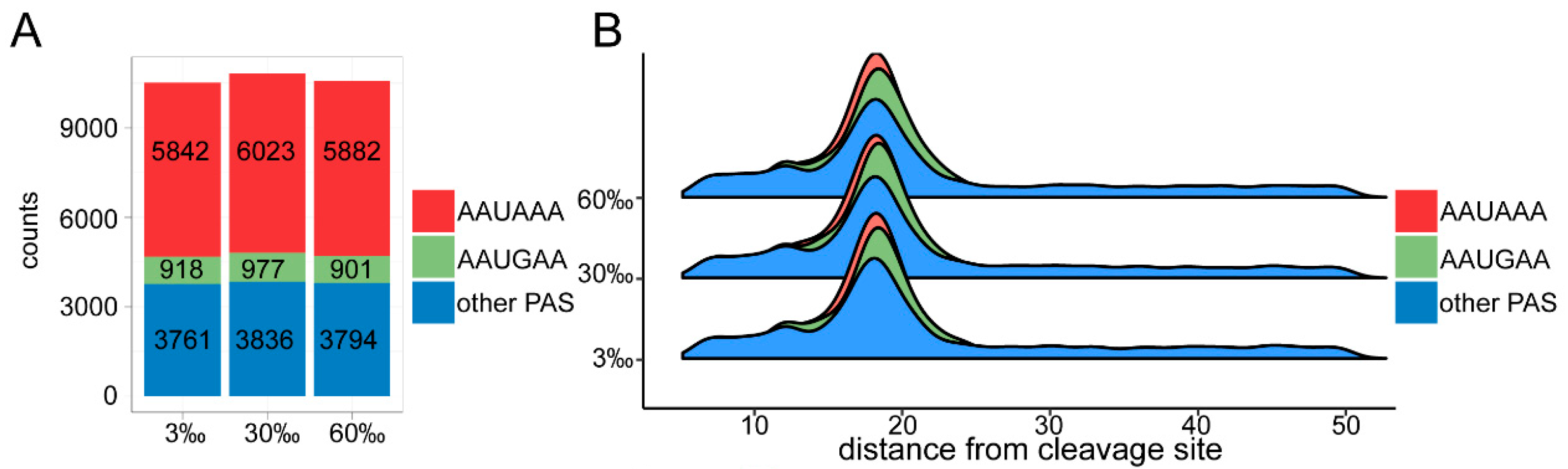
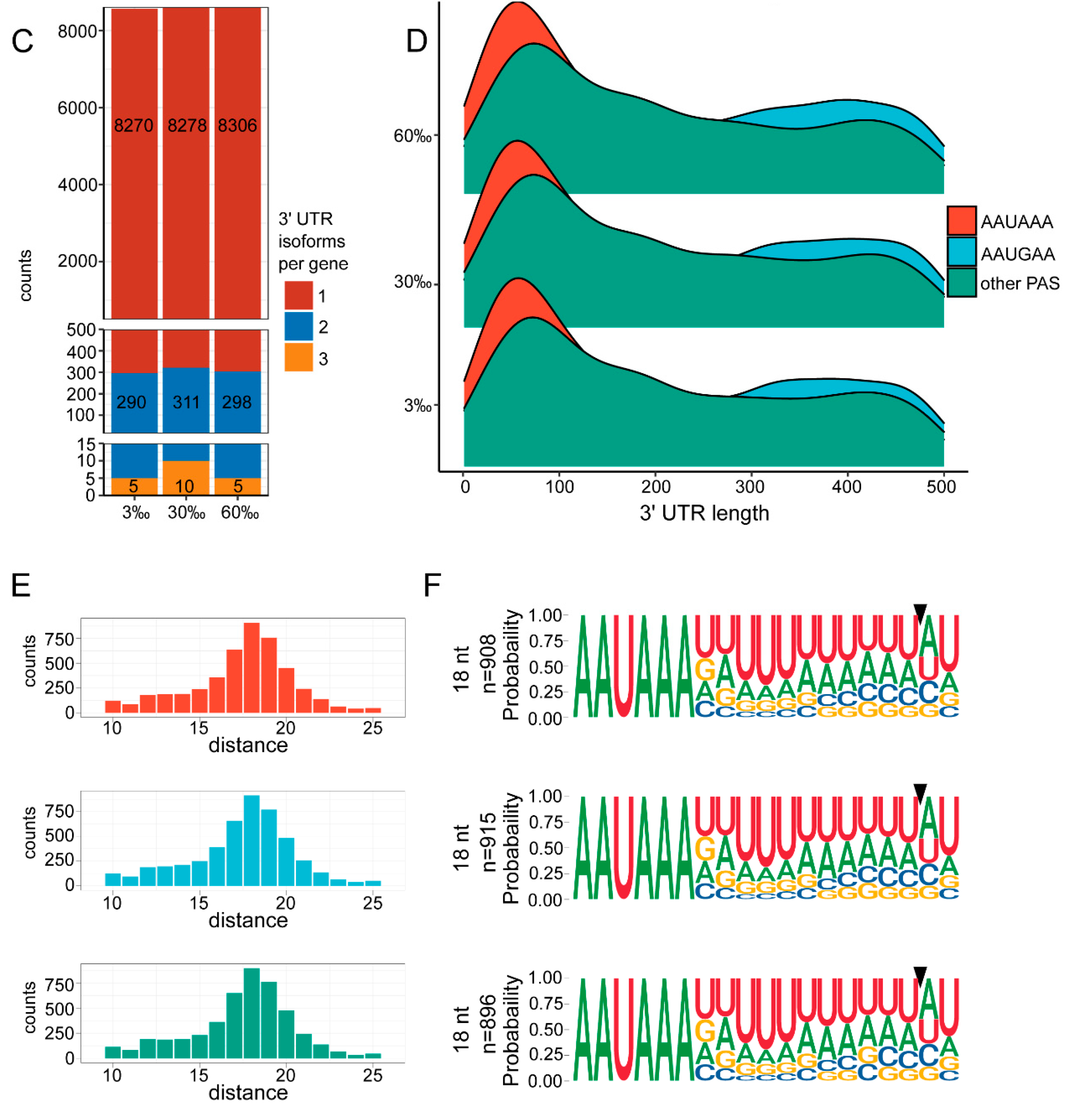
3.4. Landscape of 3′ UTRs
3.5. APA Analysis
4. Discussion
4.1. DEGs from ONT lrRNA-seq Are Highly Correlated with Illumina Data
4.2. Upregulated and Downregulated DEGs When Salinity Increased, Revealed by lrRNA–seq
4.3. DAS Identification under Different Salinity Conditions
4.4. The Marine Nematode Exhibits Distinct APA Usage under Different Salinity Conditions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xie, Y.; Zhang, P.; Zhang, L. Genome-Wide Transcriptional Responses of Marine Nematode Litoditis marina to Hyposaline and Hypersaline Stresses. Front. Physiol. 2021, 12, 672099. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Du, M.R.M.; Gouil, Q.; Tian, L.; Jabbari, J.S.; Bowden, R.; Baldoni, P.L.; Chen, Y.; Smyth, G.K.; Amarasinghe, S.L.; et al. Benchmarking long-read RNA-sequencing analysis tools using in silico mixtures. Nat. Methods 2023, 20, 1810–1821. [Google Scholar] [CrossRef] [PubMed]
- Mestre-Tomás, J.; Liu, T.; Pardo-Palacios, F.; Conesa, A. SQANTI-SIM: A simulator of controlled transcript novelty for lrRNA-seq benchmark. Genome Biol. 2023, 24, 286. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lu, Y.; Duan, Z.; Wu, J.; Lin, M.; Wu, Y.; Han, S.; Li, T.; Fan, Y.; Hu, X.; et al. Nanopore long-read RNA sequencing reveals functional alternative splicing variants in human vascular smooth muscle cells. Commun. Biol. 2023, 6, 1104. [Google Scholar] [CrossRef]
- Zhang, Z.; Bae, B.; Cuddleston, W.H.; Miura, P. Coordination of alternative splicing and alternative polyadenylation revealed by targeted long read sequencing. Nat. Commun. 2023, 14, 5506. [Google Scholar] [CrossRef] [PubMed]
- Glinos, D.A.; Garborcauskas, G.; Hoffman, P.; Ehsan, N.; Jiang, L.; Gokden, A.; Dai, X.; Aguet, F.; Brown, K.L.; Garimella, K.; et al. Transcriptome variation in human tissues revealed by long-read sequencing. Nature 2022, 608, 353–359. [Google Scholar] [CrossRef]
- Montañés, J.C.; Huertas, M.; Moro, S.G.; Blevins, W.R.; Carmona, M.; Ayté, J.; Hidalgo, E.; Albà, M.M. Native RNA sequencing in fission yeast reveals frequent alternative splicing isoforms. Genome Res. 2022, 32, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.V.; Judd, L.M.; Jex, A.R.; Holt, K.E.; Tonkin, C.J.; Ralph, S.A. Direct Nanopore Sequencing of mRNA Reveals Landscape of Transcript Isoforms in Apicomplexan Parasites. mSystems 2021, 6, e01081-20. [Google Scholar] [CrossRef]
- Gao, J.; Xu, G.C.; Xu, P. Full-length transcriptomic analysis reveals osmoregulatory mechanisms in Coilia nasus eyes reared under hypotonic and hyperosmotic stress. Sci. Total Environ. 2021, 799, 11. [Google Scholar] [CrossRef]
- Gao, J.; Xu, G.; Xu, P. Gills full-length transcriptomic analysis of osmoregulatory adaptive responses to salinity stress in Coilia nasus. Ecotoxicol. Environ. Saf. 2021, 226, 112848. [Google Scholar] [CrossRef]
- Du, Y.; Cao, L.; Wang, S.; Guo, L.; Tan, L.; Liu, H.; Feng, Y.; Wu, W. Differences in alternative splicing and their potential underlying factors between animals and plants. J. Adv. Res. 2023; in press. [Google Scholar] [CrossRef]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef]
- Wang, J.; Ma, X.; Hu, Y.; Feng, G.; Guo, C.; Zhang, X.; Ma, H. Regulation of micro- and small-exon retention and other splicing processes by GRP20 for flower development. Nat. Plants 2024, 10, 66–85. [Google Scholar] [CrossRef]
- Torre, D.; Francoeur, N.J.; Kalma, Y.; Gross Carmel, I.; Melo, B.S.; Deikus, G.; Allette, K.; Flohr, R.; Fridrikh, M.; Vlachos, K.; et al. Isoform-resolved transcriptome of the human preimplantation embryo. Nat. Commun. 2023, 14, 6902. [Google Scholar] [CrossRef]
- Liu, Z.; Zhu, C.; Steinmetz, L.M.; Wei, W. Identification and quantification of small exon-containing isoforms in long-read RNA sequencing data. Nucleic Acids. Res. 2023, 51, e104. [Google Scholar] [CrossRef]
- Ye, X.; He, C.; Yang, Y.; Sun, Y.H.; Xiong, S.; Chan, K.C.; Si, Y.; Xiao, S.; Zhao, X.; Lin, H.; et al. Comprehensive isoform-level analysis reveals the contribution of alternative isoforms to venom evolution and repertoire diversity. Genome Res. 2023, 33, 1554–1567. [Google Scholar] [CrossRef]
- Ray, M.; Conard, A.M.; Urban, J.; Mahableshwarkar, P.; Aguilera, J.; Huang, A.; Vaidyanathan, S.; Larschan, E. Sex-specific splicing occurs genome-wide during early Drosophila embryogenesis. Elife 2023, 12, e87865. [Google Scholar] [CrossRef] [PubMed]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 40. [Google Scholar] [CrossRef] [PubMed]
- Nurmi, J.T.; Puolakkainen, P.A.; Rautonen, N.E. Intron 1 retaining cyclooxygenase 1 splice variant is induced by osmotic stress in human intestinal epithelial cells. Prostaglandins Leukot Essent Fat. Acids 2005, 73, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, H.; Shenkar, N.; Zhan, A. Multidimensional plasticity jointly contributes to rapid acclimation to environmental challenges during biological invasions. RNA 2023, 29, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C. What Are 3’ UTRs Doing? Cold Spring Harb Perspect. Biol. 2019, 11, a034728. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, P.; Xue, B.; Cao, X.; Ren, X.; Wang, L.; Sun, Y.; Yang, H.; Zhang, L. Establishment of a marine nematode model for animal functional genomics, environmental adaptation and developmental evolution. bioRxiv 2020. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, L. Transcriptomic and Proteomic Analysis of Marine Nematode Litoditis marina Acclimated to Different Salinities. Genes 2022, 13, 651. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.D.; Mikheenko, A.; Joglekar, A.; Smetanin, A.; Jarroux, J.; Lapidus, A.L.; Tilgner, H.U. Accurate isoform discovery with IsoQuant using long reads. Nat. Biotechnol. 2023, 41, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Res 2020, 9. [Google Scholar] [CrossRef]
- Tardaguila, M.; de la Fuente, L.; Marti, C.; Pereira, C.; Pardo-Palacios, F.J.; Del Risco, H.; Ferrell, M.; Mellado, M.; Macchietto, M.; Verheggen, K.; et al. Corrigendum: SQANTI: Extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res. 2018, 28, 1096. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1524. [Google Scholar] [CrossRef]
- Chen, Y.; Lun, A.T.L.; Smyth, G.K. From reads to genes to pathways: Differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000research 2016, 5, 1438. [Google Scholar]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Ye, C.; Long, Y.; Ji, G.; Li, Q.Q.; Wu, X. APAtrap: Identification and quantification of alternative polyadenylation sites from RNA-seq data. Bioinformatics 2018, 34, 1841–1849. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, C.J.; Schmidt, R.; Kanitz, A.; Artimo, P.; Gruber, A.J.; Zavolan, M. PolyASite 2.0: A consolidated atlas of polyadenylation sites from 3’ end sequencing. Nucleic Acids. Res. 2020, 48, D174–D179. [Google Scholar] [CrossRef] [PubMed]
- Steber, H.S.; Gallante, C.; O’Brien, S.; Chiu, P.L.; Mangone, M. The C. elegans 3’ UTRome v2 resource for studying mRNA cleavage and polyadenylation, 3’-UTR biology, and miRNA targeting. Genome Res. 2019, 29, 2104–2116. [Google Scholar] [CrossRef]
- Hahn-Windgassen, A.; Van Gilst, M.R. The Caenorhabditis elegans HNF4alpha Homolog, NHR-31, mediates excretory tube growth and function through coordinate regulation of the vacuolar ATPase. PLoS Genet. 2009, 5, e1000553. [Google Scholar] [CrossRef]
- Lamitina, S.T.; Strange, K. Transcriptional targets of DAF-16 insulin signaling pathway protect C. elegans from extreme hypertonic stress. Am. J. Physiol. Cell Physiol. 2005, 288, C467–G474. [Google Scholar] [CrossRef]
- Tan, D.J.; Mitra, M.; Chiu, A.M.; Coller, H.A. Intron retention is a robust marker of intertumoral heterogeneity in pancreatic ductal adenocarcinoma. NPJ Genom. Med. 2020, 5, 55. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ito, M.; Sun, S.; Chida, T.; Nakashima, K.; Suzuki, T. LUC7L3/CROP inhibits replication of hepatitis B virus via suppressing enhancer II/basal core promoter activity. Sci. Rep. 2016, 6, 36741. [Google Scholar] [CrossRef]
- Ouyang, Y.; Xia, K.; Yang, X.; Zhang, S.; Wang, L.; Ren, S.; Zhou, H.; Liu, Y.; Tang, F. Alternative splicing acts as an independent prognosticator in ovarian carcinoma. Sci. Rep. 2021, 11, 10413. [Google Scholar] [CrossRef]
- Kaletsky, R.; Lakhina, V.; Arey, R.; Williams, A.; Landis, J.; Ashraf, J.; Murphy, C.T. The C. elegans adult neuronal IIS/FOXO transcriptome reveals adult phenotype regulators. Nature 2016, 529, 92–96. [Google Scholar] [CrossRef]
- Chin, R.M.; Fu, X.; Pai, M.Y.; Vergnes, L.; Hwang, H.; Deng, G.; Diep, S.; Lomenick, B.; Meli, V.S.; Monsalve, G.C.; et al. The metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014, 510, 397–401. [Google Scholar] [CrossRef]
- Johnson, L.C.; Vo, A.A.; Clancy, J.C.; Myles, K.M.; Pooranachithra, M.; Aguilera, J.; Levenson, M.T.; Wohlenberg, C.; Rechtsteiner, A.; Ragle, J.M.; et al. NHR-23 activity is necessary for C. elegans developmental progression and apical extracellular matrix structure and function. Development 2023, 150, dev201085. [Google Scholar] [CrossRef]
- De Jong, L.; Meng, Y.; Dent, J.; Hekimi, S. Thiamine pyrophosphate biosynthesis and transport in the nematode Caenorhabditis elegans. Genetics 2004, 168, 845–854. [Google Scholar] [CrossRef]
- Tang, P.; Yang, Y.; Li, G.; Huang, L.; Wen, M.; Ruan, W.; Guo, X.; Zhang, C.; Zuo, X.; Luo, D.; et al. Alternative polyadenylation by sequential activation of distal and proximal PolyA sites. Nat. Struct. Mol. Biol. 2022, 29, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Shulman, E.D.; Elkon, R. Systematic identification of functional SNPs interrupting 3’UTR polyadenylation signals. PLoS Genet. 2020, 16, e1008977. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Freitas, J.; Zheng, D.; Oliveira, M.S.; Hoque, M.; Martins, T.; Henriques, T.; Tian, B.; Moreira, A. Transcription elongation rate has a tissue-specific impact on alternative cleavage and polyadenylation in Drosophila melanogaster. RNA 2017, 23, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Chen, D.; Qi, Z.; Wang, H.; Chen, Y.; Wang, Q.; Jiang, C.; Xu, J.R.; Liu, H. Landscape and regulation of alternative splicing and alternative polyadenylation in a plant pathogenic fungus. New Phytol. 2022, 235, 674–689. [Google Scholar] [CrossRef]
- Li, W.; Park, J.Y.; Zheng, D.; Hoque, M.; Yehia, G.; Tian, B. Alternative cleavage and polyadenylation in spermatogenesis connects chromatin regulation with post-transcriptional control. BMC Biol. 2016, 14, 6. [Google Scholar] [CrossRef]
- Ji, Z.; Lee, J.Y.; Pan, Z.; Jiang, B.; Tian, B. Progressive lengthening of 3’ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc. Natl. Acad. Sci. USA 2009, 106, 7028–7033. [Google Scholar] [CrossRef]
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating cells express mRNAs with shortened 3’ untranslated regions and fewer microRNA target sites. Science 2008, 320, 1643–1647. [Google Scholar] [CrossRef]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, X.; Lei, W.; Liang, J.; Xu, Y.; Liu, H.; Ma, S. Genome-wide profiling reveals alternative polyadenylation of mRNA in human non-small cell lung cancer. J. Transl. Med. 2019, 17, 257. [Google Scholar] [CrossRef]
- Matoulkova, E.; Sommerova, L.; Pastorek, M.; Vojtesek, B.; Hrstka, R. Regulation of AGR2 expression via 3’UTR shortening. Exp. Cell Res. 2017, 356, 40–47. [Google Scholar] [CrossRef]
- Lin, A.; Ji, P.; Niu, X.; Zhao, X.; Chen, Y.; Liu, W.; Liu, Y.; Fan, W.; Sun, Y.; Miao, C.; et al. CstF64-Induced Shortening of the BID 3’UTR Promotes Esophageal Squamous Cell Carcinoma Progression by Disrupting ceRNA Cross-talk with ZFP36L2. Cancer Res. 2021, 81, 5638–5651. [Google Scholar] [CrossRef]
- Akman, H.B.; Oyken, M.; Tuncer, T.; Can, T.; Erson-Bensan, A.E. 3’UTR shortening and EGF signaling: Implications for breast cancer. Hum. Mol. Genet 2015, 24, 6910–6920. [Google Scholar] [CrossRef]
- Li, L.; Wang, D.; Xue, M.; Mi, X.; Liang, Y.; Wang, P. 3’UTR shortening identifies high-risk cancers with targeted dysregulation of the ceRNA network. Sci. Rep. 2014, 4, 5406. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, S.; Molesworth, L.W.; Gourley, T.E.; Boag, P.R.; Davis, G.M. Zinc transporters maintain longevity by influencing insulin/IGF-1 activity in Caenorhabditis elegans. Febs Lett. 2020, 594, 1424–1432. [Google Scholar] [CrossRef]
- Mootz, D.; Ho, D.M.; Hunter, C.P. The STAR/Maxi-KH domain protein GLD-1 mediates a developmental switch in the translational control of C. elegans PAL-1. Development 2004, 131, 3263–3272. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Million Reads | Mean Length | Number of Isoform/Gene | Number of Novel Isoform/Gene | |
|---|---|---|---|---|
| 3‰ | 5.34 | 737.80 | 13,769/10,985 | 3161/2549 |
| 30‰ | 5.01 | 903.60 | 14,098/10,954 | 3538/2800 |
| 60‰ | 6.25 | 753.91 | 13,920/11,083 | 3226/2594 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, P.; Xue, B.; Yang, H.; Zhang, L. Transcriptome Responses to Different Salinity Conditions in Litoditis marina, Revealed by Long-Read Sequencing. Genes 2024, 15, 317. https://doi.org/10.3390/genes15030317
Zhang P, Xue B, Yang H, Zhang L. Transcriptome Responses to Different Salinity Conditions in Litoditis marina, Revealed by Long-Read Sequencing. Genes. 2024; 15(3):317. https://doi.org/10.3390/genes15030317
Chicago/Turabian StyleZhang, Pengchi, Beining Xue, Hanwen Yang, and Liusuo Zhang. 2024. "Transcriptome Responses to Different Salinity Conditions in Litoditis marina, Revealed by Long-Read Sequencing" Genes 15, no. 3: 317. https://doi.org/10.3390/genes15030317
APA StyleZhang, P., Xue, B., Yang, H., & Zhang, L. (2024). Transcriptome Responses to Different Salinity Conditions in Litoditis marina, Revealed by Long-Read Sequencing. Genes, 15(3), 317. https://doi.org/10.3390/genes15030317