
Reconstruction of Metabolic–Protein Interaction Integrated Network of Eriocheir sinensis and Analysis of Ecdysone Synthesis
 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Transformation of the Metabolic Network to a Reaction Graph
2.2. Addition of the Biomass Equation
2.3. Reconstruction of Integrated Network
2.4. Analysis of the Ecdysone Synthesis Pathway
2.4.1. Addition of the Ecdysone Synthesis Pathway
2.4.2. Determination of Biomass Synthesis Rates
2.4.3. Identification of Metabolic Reactions and Proteins That Affect the Ecdysone Synthesis
2.5. Analysis of Key Metabolic Reactions and Proteins Affecting Ecdysone Synthesis
3. Results and Discussions
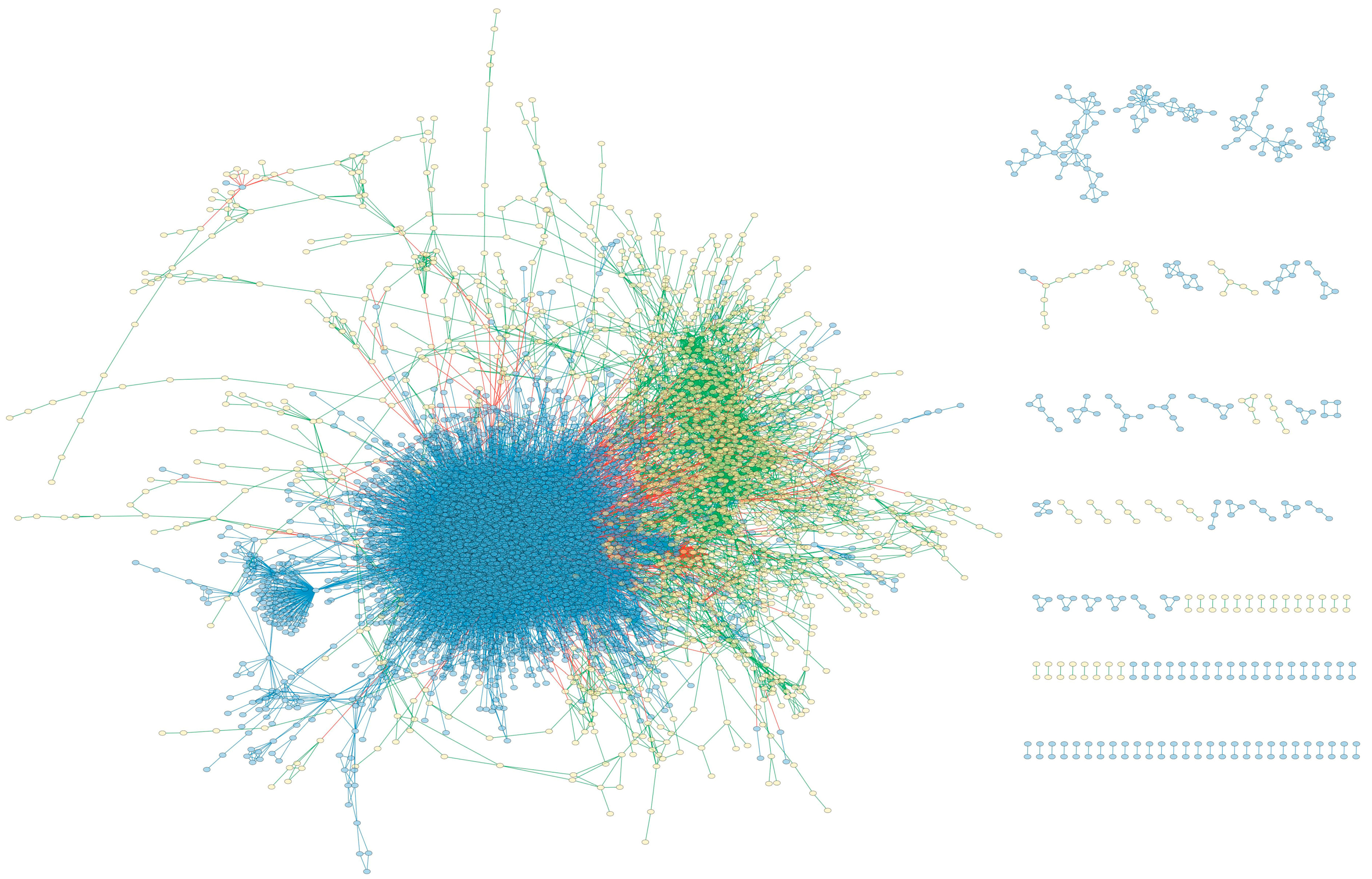
3.1. The Reconstruction of MPIN
3.1.1. Transformation of Metabolic Networks and Addition of Biomass Equations
3.1.2. Preliminary Reconstruction of Integrated Network
3.1.3. Addition of Biomass Equation
3.1.4. Correction of the Model
3.2. Analysis of the Ecdysteroid Pathway
3.2.1. Addition of the Ecdysone Synthesis Pathway
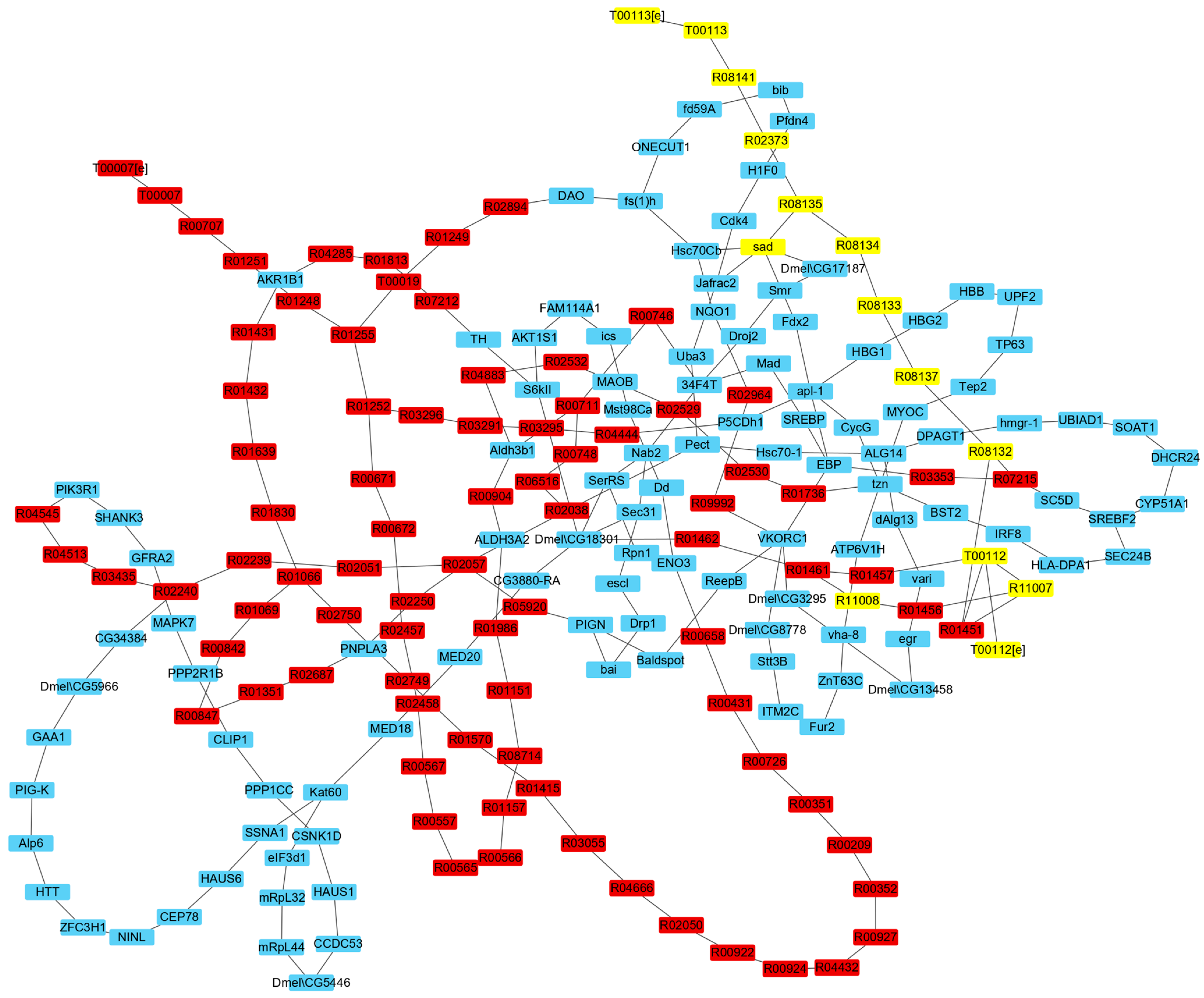
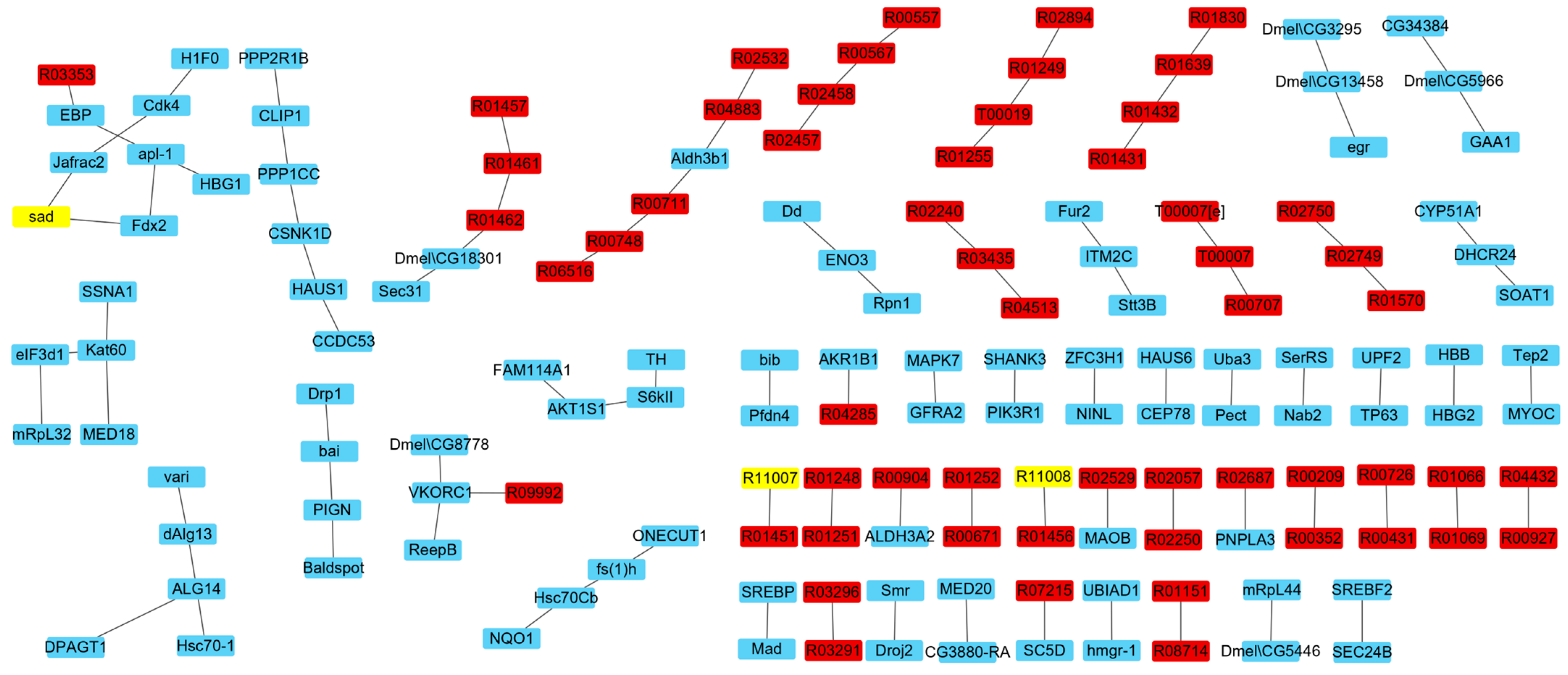
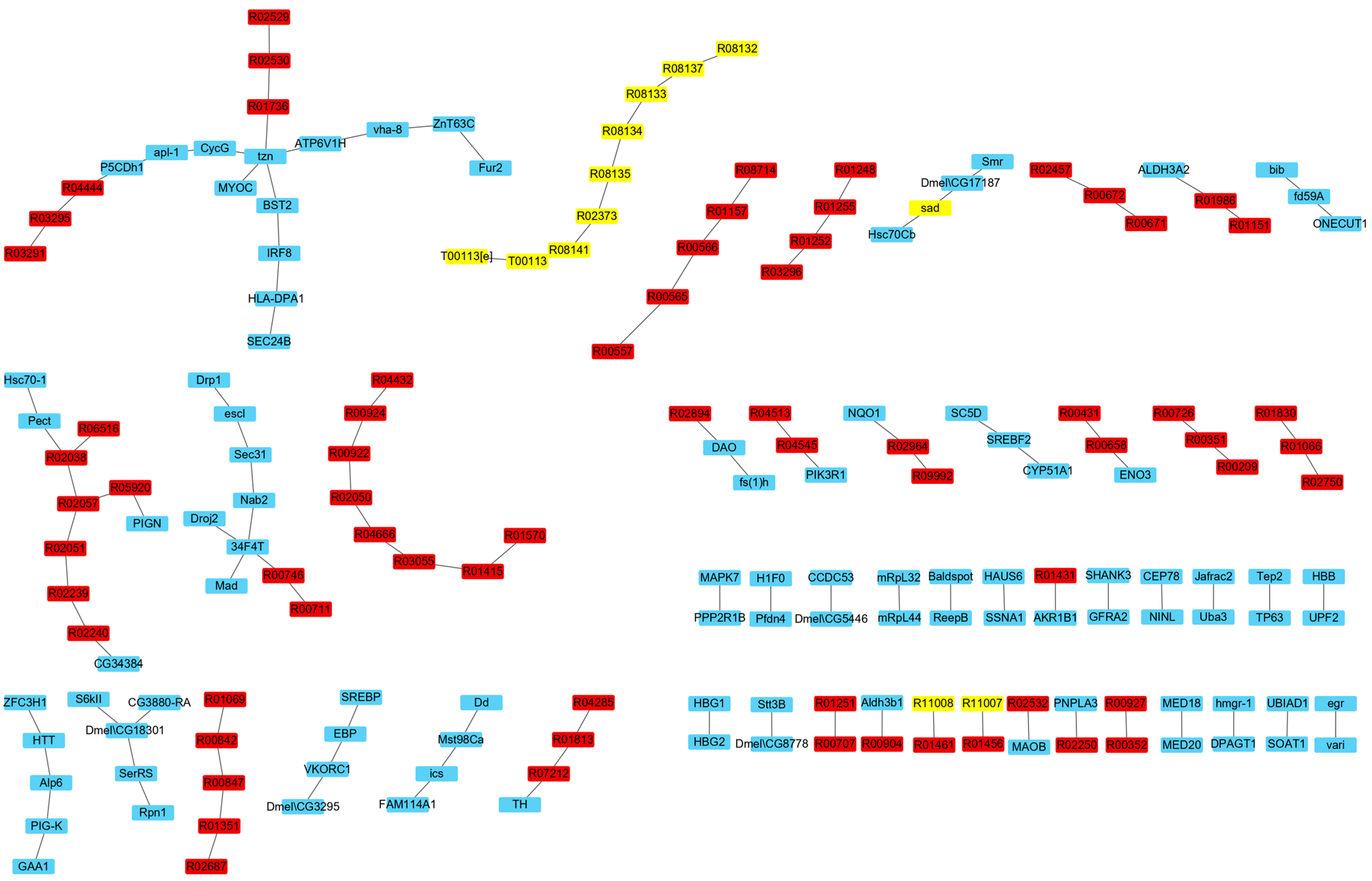
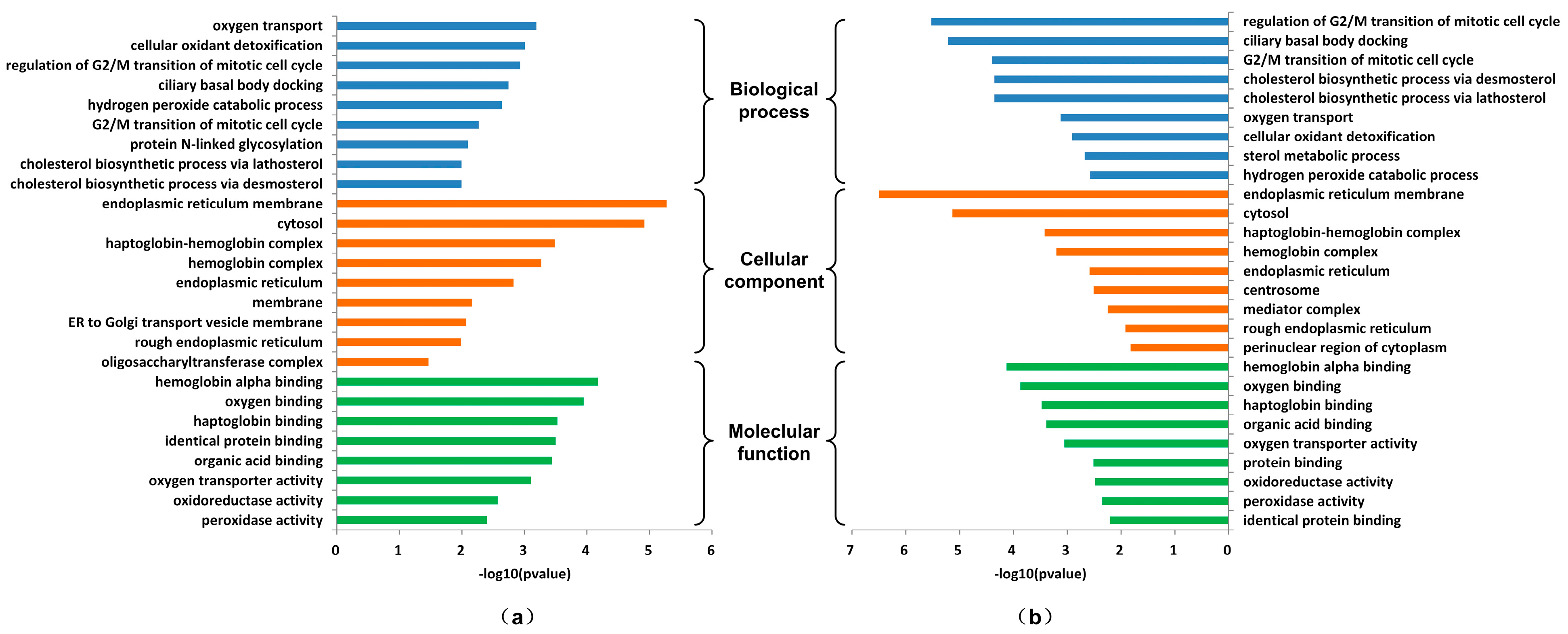
3.2.2. IRs Affecting the Synthesis of Ecdysone
3.3. Analysis of Reactions and Proteins Affecting Ecdysone Synthesis
3.3.1. Analysis of Reactions Affecting the Synthesis of Ecdysone
3.3.2. Analysis of Proteins Affecting Ecdysone Synthesis
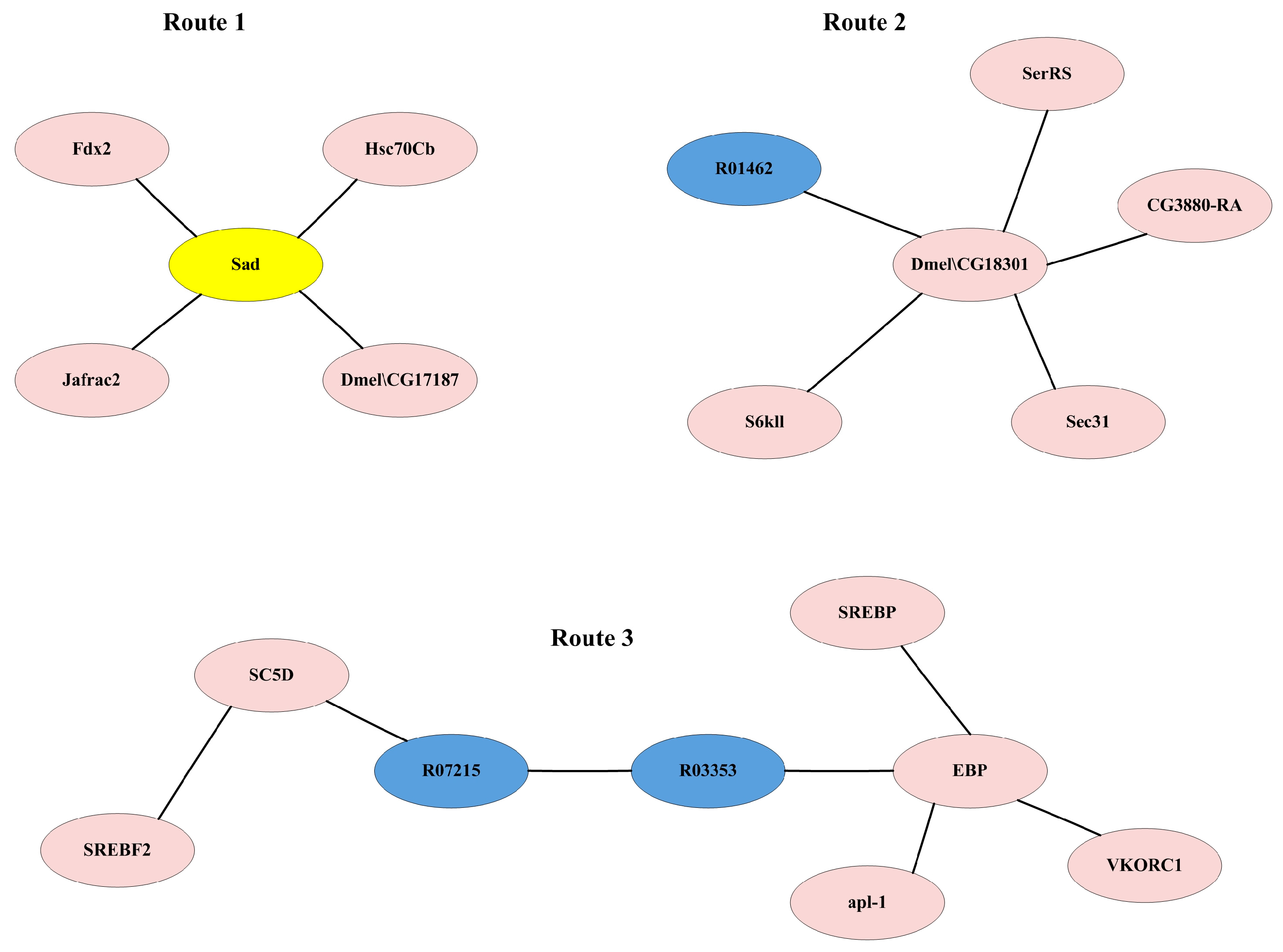
3.3.3. Analysis of Key Reactions and Proteins Affecting Ecdysone Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hao, T.; Wu, D.; Zhao, L.; Wang, Q.; Wang, E.; Sun, J. The Genome-Scale Integrated Networks in Microorganisms. Front. Microbiol. 2018, 9, 296. [Google Scholar] [CrossRef]
- Covert, M.W.; Knight, E.M.; Reed, J.L.; Herrgard, M.J.; Palsson, B.O. Integrating high-throughput and computational data elucidates bacterial networks. Nature 2004, 429, 92–96. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Price, N.D. Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 17845–17850. [Google Scholar] [CrossRef]
- Jiang, J.; Song, L.; Zheng, P.; Jia, S.; Sun, J. Construction and structural analysis of integrated cellular network of Corynebacterium glutamicum. Chin. J. Biotechnol. 2012, 28, 577–591. [Google Scholar]
- Jensen, P.A.; Lutz, K.A.; Papin, J.A. TIGER: Toolbox for integrating genome-scale metabolic models, expression data, and transcriptional regulatory networks. BMC Syst. Biol. 2011, 5, 147. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, S. A Protocol for the Construction and Curation of Genome-Scale Integrated Metabolic and Regulatory Network Models. Methods Mol. Biol. 2019, 1927, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Chen, B.S. Integrated cellular network of transcription regulations and protein-protein interactions. BMC Syst. Biol. 2010, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Covert, M.W.; Xiao, N.; Chen, T.J.; Karr, J.R. Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics 2008, 24, 2044–2050. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Gianchandani, E.P.; Eddy, J.A.; Papin, J.A. Dynamic analysis of integrated signaling, metabolic, and regulatory networks. PLoS Comput. Biol. 2008, 4, e1000086. [Google Scholar] [CrossRef]
- Karr, J.R.; Sanghvi, J.C.; Macklin, D.N.; Gutschow, M.V.; Jacobs, J.M.; Bolival, B., Jr.; Assad-Garcia, N.; Glass, J.I.; Covert, M.W. A whole-cell computational model predicts phenotype from genotype. Cell 2012, 150, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Sanghvi, J.C.; Regot, S.; Carrasco, S.; Karr, J.R.; Gutschow, M.V.; Bolival, B., Jr.; Covert, M.W. Accelerated discovery via a whole-cell model. Nat. Methods 2013, 10, 1192–1195. [Google Scholar] [CrossRef]
- Carrera, J.; Estrela, R.; Luo, J.; Rai, N.; Tsoukalas, A.; Tagkopoulos, I. An integrative, multi-scale, genome-wide model reveals the phenotypic landscape of Escherichia coli. Mol. Syst. Biol. 2014, 10, 735. [Google Scholar] [CrossRef]
- Macklin, D.N.; Ahn-Horst, T.A.; Choi, H.; Ruggero, N.A.; Carrera, J.; Mason, J.C.; Sun, G.; Agmon, E.; DeFelice, M.M.; Maayan, I.; et al. Simultaneous cross-evaluation of heterogeneous E. coli datasets via mechanistic simulation. Science 2020, 369, aav3751. [Google Scholar] [CrossRef]
- Sun, G.; Ahn-Horst, T.A.; Covert, M.W. The E. coli Whole-Cell Modeling Project. EcoSal Plus 2021, 9, eESP00012020. [Google Scholar] [CrossRef]
- Ahn-Horst, T.A.; Mille, L.S.; Sun, G.; Morrison, J.H.; Covert, M.W. An expanded whole-cell model of E. coli links cellular physiology with mechanisms of growth rate control. npj Syst. Biol. Appl. 2022, 8, 30. [Google Scholar] [CrossRef]
- Hao, T.; Peng, W.; Wang, Q.; Wang, B.; Sun, J. Reconstruction and Application of Protein-Protein Interaction Network. Int. J. Mol. Sci. 2016, 17, 907. [Google Scholar] [CrossRef]
- Wang, B.; Yang, J.; Gao, C.; Hao, T.; Li, J.; Sun, J. Reconstruction of Eriocheir sinensis Y-organ Genome-Scale Metabolic Network and Differential Analysis After Eyestalk Ablation. Front. Genet. 2020, 11, 532492. [Google Scholar] [CrossRef]
- Chen, X.; Wang, J.; Hou, X.; Yue, W.; Huang, S.; Wang, C. Tissue expression profiles unveil the gene interaction of hepatopancreas, eyestalk, and ovary in the precocious female Chinese mitten crab, Eriocheir sinensis. BMC Genet. 2019, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, J.; Yue, W.; Huang, S.; Chen, J.; Chen, Y.; Wang, C. Structure and function of the alternatively spliced isoforms of the ecdysone receptor gene in the Chinese mitten crab, Eriocheir sinensis. Sci. Rep. 2017, 7, 12993. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Yang, H.; Chen, X.; Wang, J.; Wang, C. RNA interference of mTOR gene delays molting process in Eriocheir sinensis. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2021, 256, 110651. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Lizarraga, A.E.; Juarez-Morales, J.L.; Racotta, I.S.; Villarreal-Colmenares, H.; Valdes-Lopez, O.; Luna-Gonzalez, A.; Rodriguez-Jaramillo, C.; Estrada, N.; Ascencio, F. Transcriptomic analysis of Pacific white shrimp (Litopenaeus vannamei, Boone 1931) in response to acute hepatopancreatic necrosis disease caused by Vibrio parahaemolyticus. PLoS ONE 2019, 14, e0220993. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gou, Y.; Yang, J.; Zhao, L.; Wang, B.; Hao, T.; Sun, J. Genome-scale metabolic network model of Eriocheir sinensis icrab4665 and nutritional requirement analysis. BMC Genom. 2022, 23, 475. [Google Scholar] [CrossRef] [PubMed]
- Hao, T.; Gou, Y.; Li, J.; Wang, B.; Zhang, Y.; Sun, J. Construction of Eriocheir sinensis Protein-protein Interaction Network and Extraction of Molting Sub-network. In Proceedings of the 12th International Conference on Bioscience, Biochemistry and Bioinformatics, Tokyo, Japan, 7–10 January 2022; pp. 70–76. [Google Scholar]
- Wang, B.; Ning, Q.; Hao, T.; Yu, A.; Sun, J. Reconstruction and analysis of a genome-scale metabolic model for Eriocheir sinensis eyestalks. Mol. Biosyst. 2016, 12, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ge, Y. GB28050-2011 General principles for nutrition labeling of prepackaged food in national food safety standard. Sci. Technol. Food Ind. 2013, 18, 24–27. [Google Scholar]
- Huang, S.; Yi, Q.; Lian, X.; Xu, S.; Yang, C.; Sun, J.; Wang, L.; Song, L. The involvement of ecdysone and ecdysone receptor in regulating the expression of antimicrobial peptides in Chinese mitten crab, Eriocheir sinensis. Dev. Comp. Immunol. 2020, 111, 103757. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Li, X.; Jiang, Y.; Si, Y.; Zheng, Y.; Sun, N. Comparative Growth Traits in Different Families of Juvenile Chinese Mitten Handed Crab, Eriocheir sinensis, Cultured in Net Cages Disposed in Ricefields and in an Indoor Tank. Fish. Sci. 2016, 35, 547–551. [Google Scholar]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdottir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Ewald, C.Y.; Raps, D.A.; Li, C. APL-1, the Alzheimer’s Amyloid precursor protein in Caenorhabditis elegans, modulates multiple metabolic pathways throughout development. Genetics 2012, 191, 493–507. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Count |
|---|---|
| Reaction | 1793 |
| Reversible-metabolic-reaction | 405 |
| Irreversible-metabolic-reaction | 1388 |
| Exchange reaction | 89 |
| Transport reaction | 89 |
| R-R interactions | 4636 |
| Reversible R-R interaction | 756 |
| Irreversible R-R interaction | 3562 |
| Reversible T-R interaction | 73 |
| Irreversible T-R interaction | 245 |
| Metabolites | 1658 |
| Item | Count |
|---|---|
| Nodes | 10,301 |
| Protein | 8225 |
| Metabolic reaction | 1793 |
| Transport reaction | 89 |
| Exchange reaction | 89 |
| IRs | 153,305 |
| P-P IR | 147,656 |
| R-R IR | 4318 |
| T-T IR | 89 |
| T-E IR | 89 |
| T-R IR | 318 |
| P-R IR | 835 |
| Item | Count |
|---|---|
| New-Nodes | 2 |
| B00001[c] | 1 |
| B00001[e] | 1 |
| New-IRs | 148 |
| R-B IR | 128 |
| T-B IR | 20 |
| T-T IR | 1 |
| T-E IR | 1 |
| Item | Number |
|---|---|
| Nodes | 10,267 |
| Protein | 8225 |
| Metabolic reaction | 1816 |
| Transport | 112 |
| Exchange | 112 |
| Biomass | 2 |
| IR | 153,611 |
| P-P IR | 147,656 |
| R-R IR | 4364 |
| T-T IR | 113 |
| T-E IR | 113 |
| T-R IR | 391 |
| P-R IR | 835 |
| R-B IR | 128 |
| T-B IR | 20 |
| Item | Number |
|---|---|
| Nodes | 10,282 |
| Protein | 8225 |
| Metabolic reaction | 1827 |
| Transport reaction | 114 |
| Exchange reaction | 114 |
| Biomass reaction | 2 |
| IR | 153,647 |
| P-P IR | 147,656 |
| R-R IR | 4376 |
| T-T IR | 115 |
| T-E IR | 115 |
| T-R IR | 396 |
| P-R IR | 841 |
| R-B IR | 128 |
| T-B IR | 20 |
| Subsystem | Pathway | Competitive Reaction | Promotive Reaction |
|---|---|---|---|
| Glucose metabolism | Glycolysis/Gluconeogenesis | 1 | 3 |
| Citrate cycle (TCA cycle) | 1 | 2 | |
| Pentose phosphate pathway | 4 | 3 | |
| Pentose and glucuronate interconversions | 3 | 1 | |
| Galactose metabolism | 1 | 1 | |
| Inositol phosphate metabolism | 2 | 2 | |
| Pyruvate metabolism | 2 | 1 | |
| Propanoate metabolism | 1 | 1 | |
| GPI-anchor biosynthesis | 0 | 1 | |
| Amino acid metabolism | Arginine biosynthesis | 1 | 1 |
| Alanine, aspartate and glutamate metabolism | 1 | 1 | |
| Glycine, serine and threonine metabolism | 1 | 1 | |
| Valine, leucine and isoleucine degradation | 1 | 3 | |
| Arginine and proline metabolism | 11 | 17 | |
| Tyrosine metabolism | 2 | 1 | |
| β-Alanine metabolism | 1 | 2 | |
| D-Amino acid metabolism | 3 | 2 | |
| Lipid metabolism | Steroid biosynthesis | 7 | 5 |
| Glycerolipid metabolism | 3 | 6 | |
| Glycerophospholipid metabolism | 2 | 4 | |
| Sphingolipid metabolism | 1 | 1 | |
| Cofactor, vitamin metabolism | Ubiquinone and other terpenoid-quinone biosynthesis | 1 | 2 |
| Folate biosynthesis | 1 | 3 | |
| Nucleic acid metabolism | Pyrimidine metabolism | 1 | 4 |
| Metabolism of Terpenoids and Ketones | Insect hormone biosynthesis | 2 | 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, T.; Song, Z.; Zhang, M.; Zhang, L.; Yang, J.; Li, J.; Sun, J. Reconstruction of Metabolic–Protein Interaction Integrated Network of Eriocheir sinensis and Analysis of Ecdysone Synthesis. Genes 2024, 15, 410. https://doi.org/10.3390/genes15040410
Hao T, Song Z, Zhang M, Zhang L, Yang J, Li J, Sun J. Reconstruction of Metabolic–Protein Interaction Integrated Network of Eriocheir sinensis and Analysis of Ecdysone Synthesis. Genes. 2024; 15(4):410. https://doi.org/10.3390/genes15040410
Chicago/Turabian StyleHao, Tong, Zhentao Song, Mingzhi Zhang, Lingrui Zhang, Jiarui Yang, Jingjing Li, and Jinsheng Sun. 2024. "Reconstruction of Metabolic–Protein Interaction Integrated Network of Eriocheir sinensis and Analysis of Ecdysone Synthesis" Genes 15, no. 4: 410. https://doi.org/10.3390/genes15040410