
Detection and Analysis of Genes Affecting the Number of Thoracic Vertebrae in Licha Black Pigs



Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal and Sample Collection
2.2. Whole Genome Resequencing
2.3. Quality Control and Reads Mapping
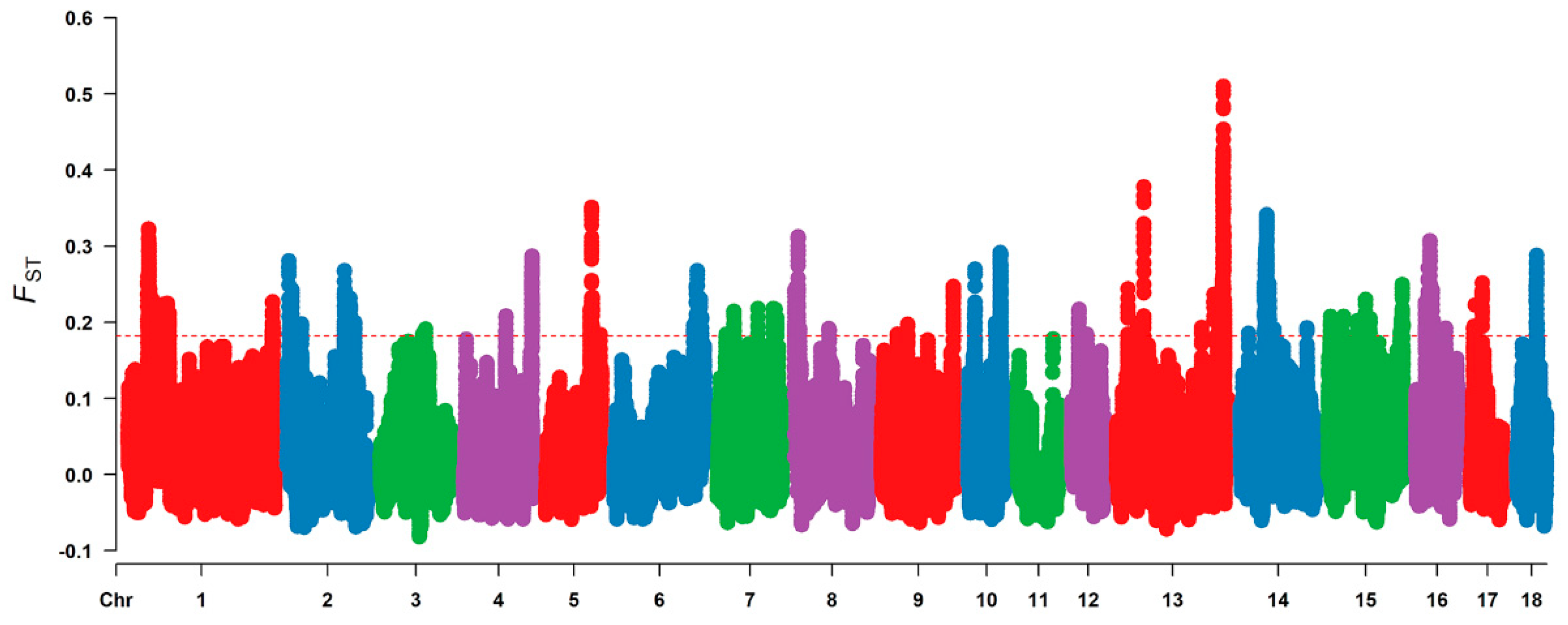
2.4. Genome Scanning for Selective Signal Analysis
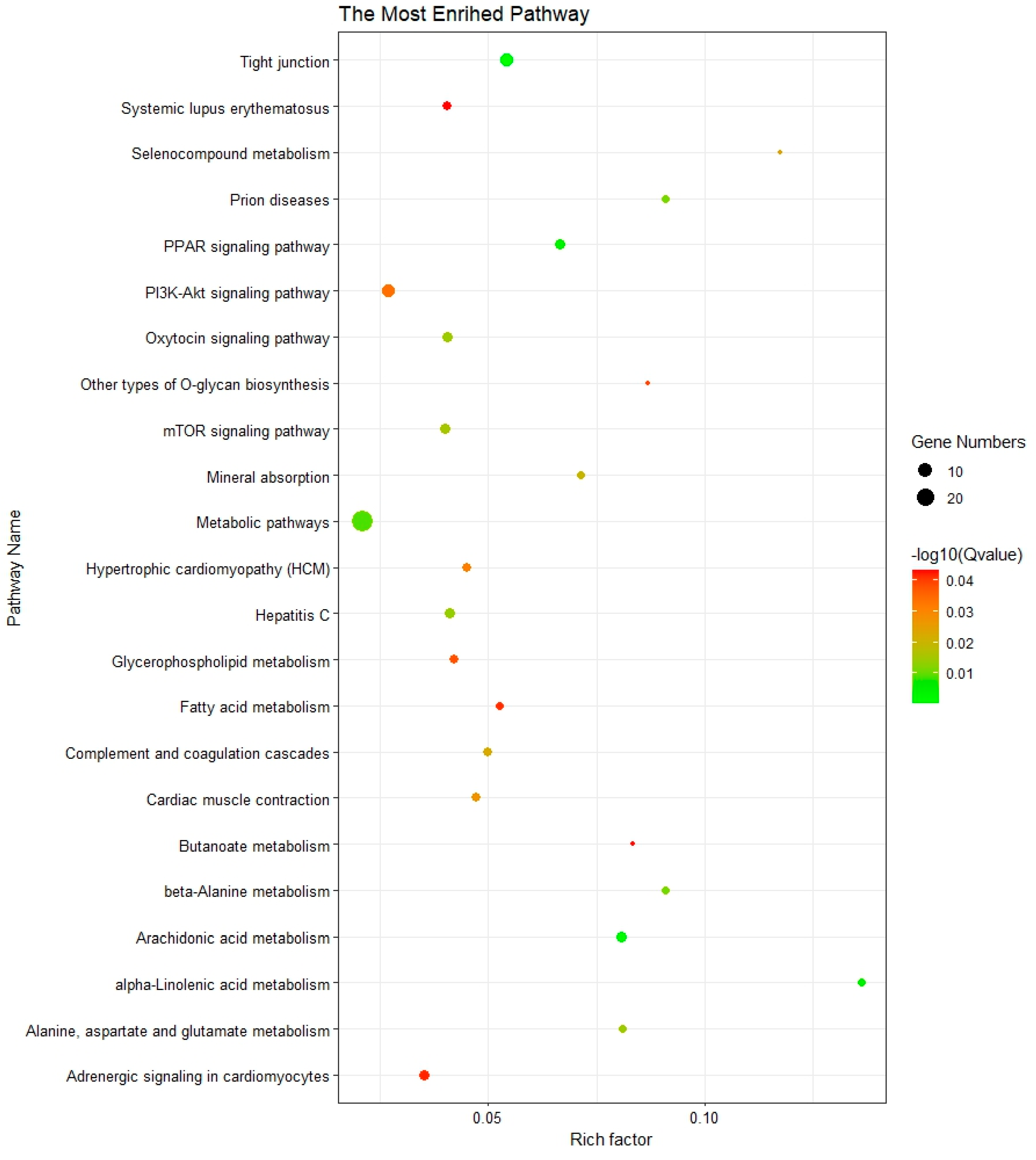
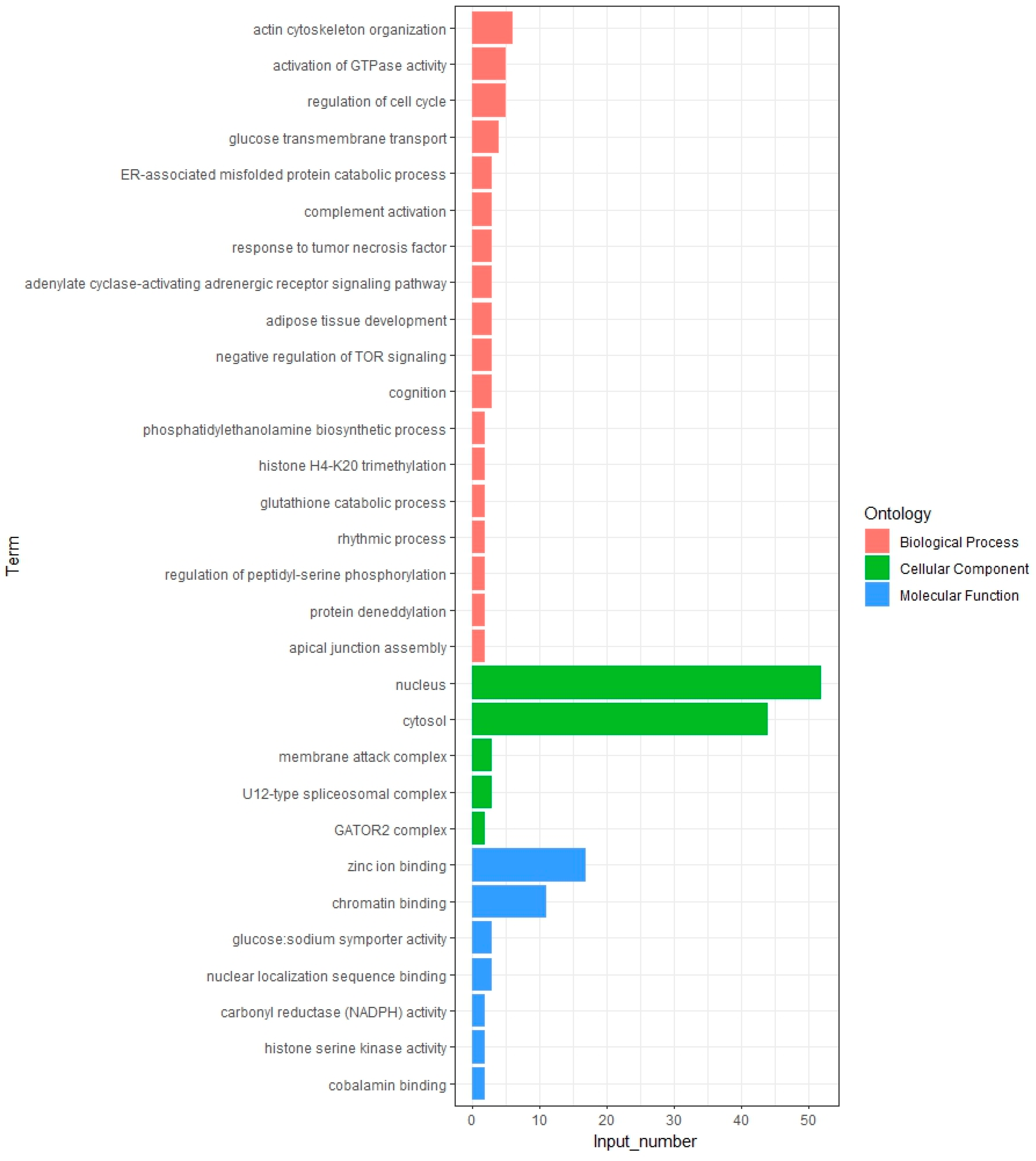
2.5. Enrichment Analysis
3. Results
3.1. Quality and Statistics of the Sequencing Data
3.2. Selective Signal Analysis
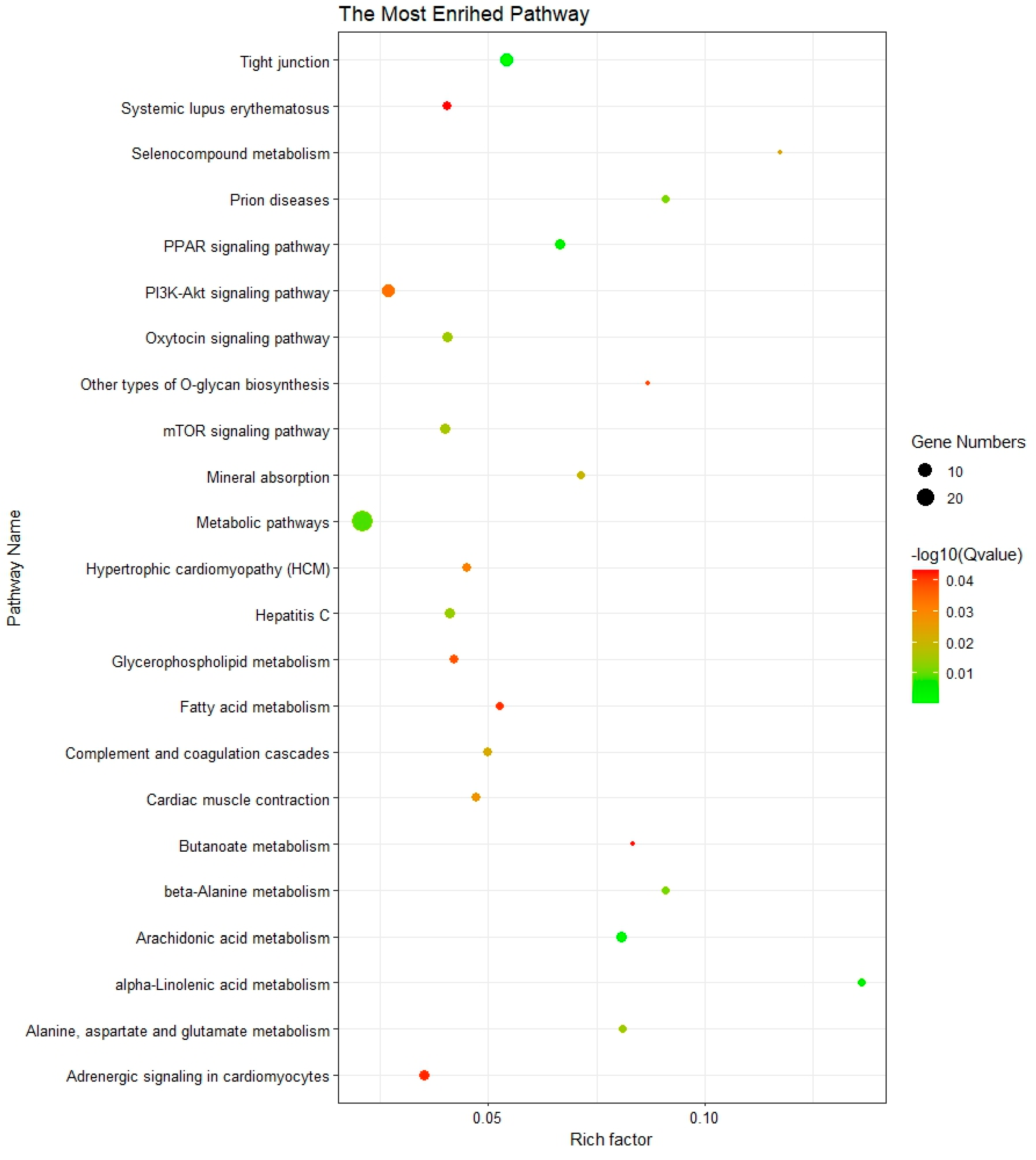
3.3. Functional Enrichment Analysis
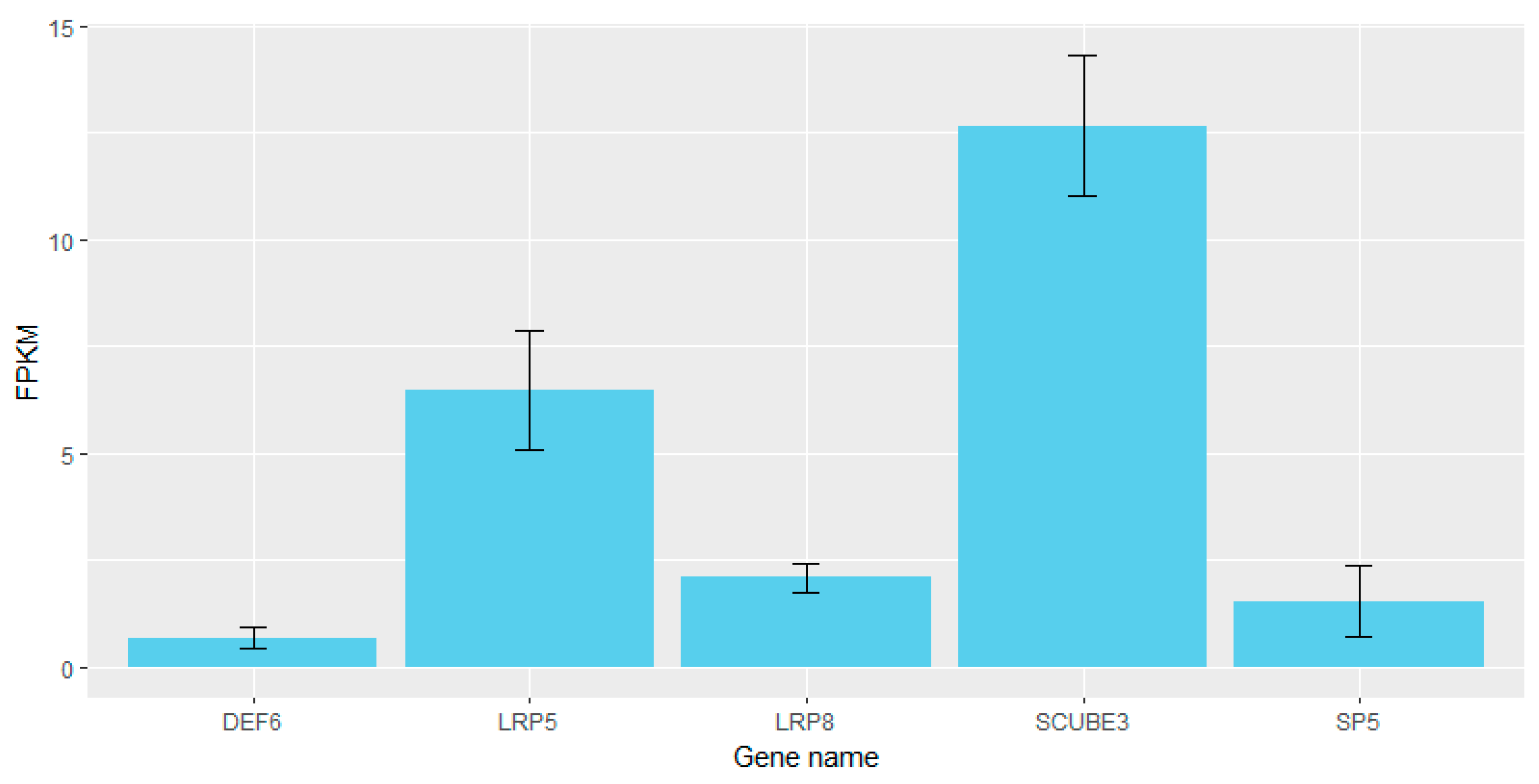
3.4. Comparison of Selective Signal Regions and QTL Database
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Borchers, N.; Reinsch, N.; Kalm, E. The number of ribs and vertebrae in a Piétrain cross: Variation, heritability and effects on performance traits. J. Anim. Breed. Genet. 2004, 121, 392–403. [Google Scholar] [CrossRef]
- King, J.W.B.; Roberts, R.C. Carcass length in the bacon pig; its association with vertebrae numbers and prediction from radiographs of the young pig. Anim. Prod. 1960, 2, 59–65. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, R.; Li, X.; Cui, C.; Yu, G. Analysis of the genetic diversity and family structure of the Licha black pig population on Jiaodong Peninsula, Shandong province, China. Animals 2022, 12, 1045. [Google Scholar] [CrossRef] [PubMed]
- Yang, H. Livestock development in China: Animal production, consumption and genetic resources. J. Anim. Breed. Genet. 2013, 130, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Ren, J.; Zhang, Z.; Huang, L. Genetic evidence for the introgression of Western NR6A1 haplotype into Chinese Licha breed associated with increased vertebral number. Anim. Genet. 2009, 40, 247–250. [Google Scholar] [CrossRef]
- van Son, M.; Lopes, M.S.; Martell, H.J.; Derks, M.F.L.; Gangsei, L.E.; Kongsro, J.; Wass, M.N.; Grindflek, E.H.; Harlizius, B. A QTL for number of teats shows breed specific effects on number of vertebrae in pigs: Bridging the gap between molecular and quantitative genetics. Front. Genet. 2019, 10, 272. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, L.; Yang, M.; Fan, Y.; Li, L.; Fang, S.; Deng, W.; Cui, L.; Zhang, Z.; Ai, H.; et al. Possible introgression of the VRTN mutation increasing vertebral number, carcass length and teat number from Chinese pigs into European pigs. Sci. Rep. 2016, 6, 19240. [Google Scholar] [CrossRef] [PubMed]
- Niu, N.; Liu, Q.; Hou, X.; Liu, X.; Wang, L.; Zhao, F.; Gao, H.; Shi, L.; Wang, L.; Zhang, L. Genome-wide association study revealed ABCD4 on SSC7 and GREB1L and MIB1 on SSC6 as crucial candidate genes for rib number in Beijing Black pigs. Anim. Genet. 2022, 53, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yue, J.; Niu, N.; Liu, X.; Yan, H.; Zhao, F.; Hou, X.; Gao, H.; Shi, L.; Wang, L.; et al. Genome-wide association analysis identified BMPR1A as a novel candidate gene affecting the number of thoracic vertebrae in a Large White x Minzhu intercross pig population. Animals 2020, 10, 2186. [Google Scholar] [CrossRef]
- Yoshioka-Kobayashi, K.; Matsumiya, M.; Niino, Y.; Isomura, A.; Kori, H.; Miyawaki, A.; Kageyama, R. Coupling delay controls synchronized oscillation in the segmentation clock. Nature 2020, 580, 119–123. [Google Scholar] [CrossRef]
- Wang, K.; Hu, H.; Tian, Y.; Li, J.; Scheben, A.; Zhang, C.; Li, Y.; Wu, J.; Yang, L.; Fan, X.; et al. The Chicken Pan-Genome Reveals Gene Content Variation and a Promoter Region Deletion in IGF2BP1 Affecting Body Size. Mol. Biol. Evol. 2021, 38, 5066–5081. [Google Scholar] [CrossRef] [PubMed]
- Dequeant, M.L.; Pourquie, O. Segmental patterning of the vertebrate embryonic axis. Nat. Rev. Genet. 2008, 9, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Rivals, I.; Personnaz, L.; Taing, L.; Potier, M.C. Enrichment or depletion of a GO category within a class of genes: Which test? Bioinformatics 2007, 23, 401–407. [Google Scholar] [CrossRef]
- Li, C.; Liu, K.; Dai, J.; Li, X.; Liu, X.; Ni, W.; Li, H.; Wang, D.; Qiao, J.; Wang, Y.; et al. Whole-genome resequencing to investigate the determinants of the multi-lumbar vertebrae trait in sheep. Gene 2022, 809, 146020. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Zhang, Z.; Chen, Z.; Sun, J.; Li, M.; Wang, Q.; Pan, Y. Integration of Selection Signatures and Protein Interactions Reveals NR6A1, PAPPA2, and PIK3C2B as the Promising Candidate Genes Underlying the Characteristics of Licha Black Pig. Biology 2023, 12, 500. [Google Scholar] [CrossRef] [PubMed]
- Balemans, W.; Piters, E.; Cleiren, E.; Ai, M.; Van Wesenbeeck, L.; Warman, M.L.; Van Hul, W. The binding between sclerostin and LRP5 is altered by DKK1 and by high-bone mass LRP5 mutations. Calcified Tissue Int. 2008, 82, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Salbach-Hirsch, J.; Samsonov, S.A.; Hintze, V.; Hofbauer, C.; Picke, A.K.; Rauner, M.; Gehrcke, J.P.; Moeller, S.; Schnabelrauch, M.; Scharnweber, D.; et al. Structural and functional insights into sclerostin-glycosaminoglycan interactions in bone. Biomaterials 2015, 67, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Zhang, D.; Cheng, F.; Wilson, D.; Mackay, J.; He, K.; Ban, Q.; Lv, F.; Huang, S.; Liu, D.; et al. Wnt/beta-catenin and LIF-Stat3 signaling pathways converge on Sp5 to promote mouse embryonic stem cell self-renewal. J. Cell Sci. 2016, 129, 269–276. [Google Scholar] [PubMed]
- Huggins, I.J.; Bos, T.; Gaylord, O.; Jessen, C.; Lonquich, B.; Puranen, A.; Richter, J.; Rossdam, C.; Brafman, D.; Gaasterland, T.; et al. The WNT target SP5 negatively regulates WNT transcriptional programs in human pluripotent stem cells. Nat. Commun. 2017, 8, 1034. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Zhang, L.; Zhou, F.; van Dinther, M.; Ten Dijke, P. LRP8 mediates Wnt/beta-catenin signaling and controls osteoblast differentiation. J. Bone Miner. Res. 2012, 27, 2065–2074. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Fanzo, J.C.; Hu, C.; Cox, D.; Jang, S.Y.; Lee, A.E.; Greenberg, S.; Pernis, A.B. T cell receptor engagement leads to the recruitment of IBP, a novel guanine nucleotide exchange factor, to the immunological synapse. J. Biol. Chem. 2003, 278, 43541–43549. [Google Scholar] [CrossRef]
- Pei, Y.F.; Liu, L.; Liu, T.L.; Yang, X.L.; Zhang, H.; Wei, X.T.; Feng, G.J.; Hai, R.; Ran, S.; Zhang, L. Joint Association Analysis Identified 18 New Loci for Bone Mineral Density. J. Bone Miner. Res. 2019, 34, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Ng, C.; Inoue, K.; Chen, Z.; Xia, Y.; Hu, X.; Greenblatt, M.; Pernis, A.; Zhao, B. Def6 regulates endogenous type-I interferon responses in osteoblasts and suppresses osteogenesis. eLife 2020, 9, e59659. [Google Scholar] [CrossRef]
- Tu, C.F.; Tsao, K.C.; Lee, S.J.; Yang, R.B. SCUBE3 (signal peptide-CUB-EGF domain-containing protein 3) modulates fibroblast growth factor signaling during fast muscle development. J. Biol. Chem. 2014, 289, 18928–18942. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Niceta, M.; Muto, V.; Vona, B.; Pagnamenta, A.T.; Maroofian, R.; Beetz, C.; van Duyvenvoorde, H.; Dentici, M.L.; Lauffer, P.; et al. SCUBE3 loss-of-function causes a recognizable recessive developmental disorder due to defective bone morphogenetic protein signaling. Am. J. Hum. Genet. 2021, 108, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Zhang, H.; Zhang, Z.; Gao, J.; Yang, J.; Wu, Z.; Fan, Y.; Xing, Y.; Li, L.; Xiao, S.; et al. VRTN is required for the development of thoracic vertebrae in mammals. Int. J. Biol. Sci. 2018, 14, 667–681. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SSC | Selection Signal Region Number | Length (Mb) 1 | Autosome Coverage (%) 2 | Gene Number 3 |
|---|---|---|---|---|
| 1 | 380 | 38 | 13.85 | 86 |
| 2 | 110 | 11 | 7.24 | 32 |
| 3 | 4 | 0.4 | 0.3 | 3 |
| 4 | 130 | 13 | 9.93 | 19 |
| 5 | 74 | 7.4 | 7.08 | 20 |
| 6 | 98 | 9.8 | 5.74 | 24 |
| 7 | 40 | 4 | 3.28 | 14 |
| 8 | 192 | 19.2 | 13.82 | 78 |
| 9 | 25 | 2.5 | 1.79 | 12 |
| 10 | 171 | 17.1 | 24.65 | 15 |
| 12 | 15 | 1.5 | 2.44 | 7 |
| 13 | 340 | 34 | 16.32 | 52 |
| 14 | 342 | 34.2 | 24.13 | 111 |
| 15 | 94 | 9.4 | 6.69 | 24 |
| 16 | 171 | 17.1 | 21.39 | 21 |
| 17 | 31 | 3.1 | 4.88 | 4 |
| 18 | 48 | 4.8 | 8.57 | 5 |
| Total 4 | 2265 | 226.5 | 10 | 527 |
| SSC | Bin Start (bp) 1 | Bin Eed (bp) | FST 2 | Gene Name 3 |
|---|---|---|---|---|
| 13 | 199,280,001 | 199,380,000 | 0.509862 | LOC106505853, LOC110256478, LOC106505851 |
| 13 | 198,830,001 | 198,930,000 | 0.425367 | LOC106505851 |
| 13 | 198,650,001 | 198,750,000 | 0.410486 | LOC106508030 |
| 13 | 198,630,001 | 198,730,000 | 0.401655 | RUNX1 |
| 13 | 50,480,001 | 50,580,000 | 0.377898 | ARL6IP5, LMOD3, LOC110256266, UBA3 |
| 13 | 50,470,001 | 50,570,000 | 0.357269 | TMF1 |
| 5 | 85,790,001 | 85,890,000 | 0.351059 | LOC102160458 |
| 13 | 199,770,001 | 199,870,000 | 0.348404 | CBR3, DOPEY2 |
| 14 | 49,210,001 | 49,310,000 | 0.34131 | BCR |
| 14 | 49,530,001 | 49,630,000 | 0.338639 | GGT1, GUCD1, LRRC75B, SNRPD3, SNRPD3, UPB1 |
| 14 | 49,550,001 | 49,650,000 | 0.335205 | GGT5 |
| 14 | 49,200,001 | 49,300,000 | 0.334916 | RAB36 |
| 14 | 49,580,001 | 49,680,000 | 0.333836 | LOC100520275, SUSD2 |
| 14 | 49,270,001 | 49,370,000 | 0.33244 | SPECC1L |
| 14 | 49,590,001 | 49,690,000 | 0.324805 | CABIN1 |
| 13 | 50,510,001 | 50,610,000 | 0.323567 | FRMD4B |
| 1 | 38,460,001 | 38,560,000 | 0.322458 | NKAIN2 |
| 13 | 199,680,001 | 199,780,000 | 0.32011 | LOC110256483 |
| 8 | 6,040,001 | 6,140,000 | 0.311989 | LOC110262054, LYAR, OTOP1, TMEM128, ZBTB49, DRD5 |
| 8 | 6,090,001 | 6,190,000 | 0.311252 | DRD5 |
| SSC | QTL Name | QTL Start (bp) 1 | QTL End (bp) | Gene Name |
|---|---|---|---|---|
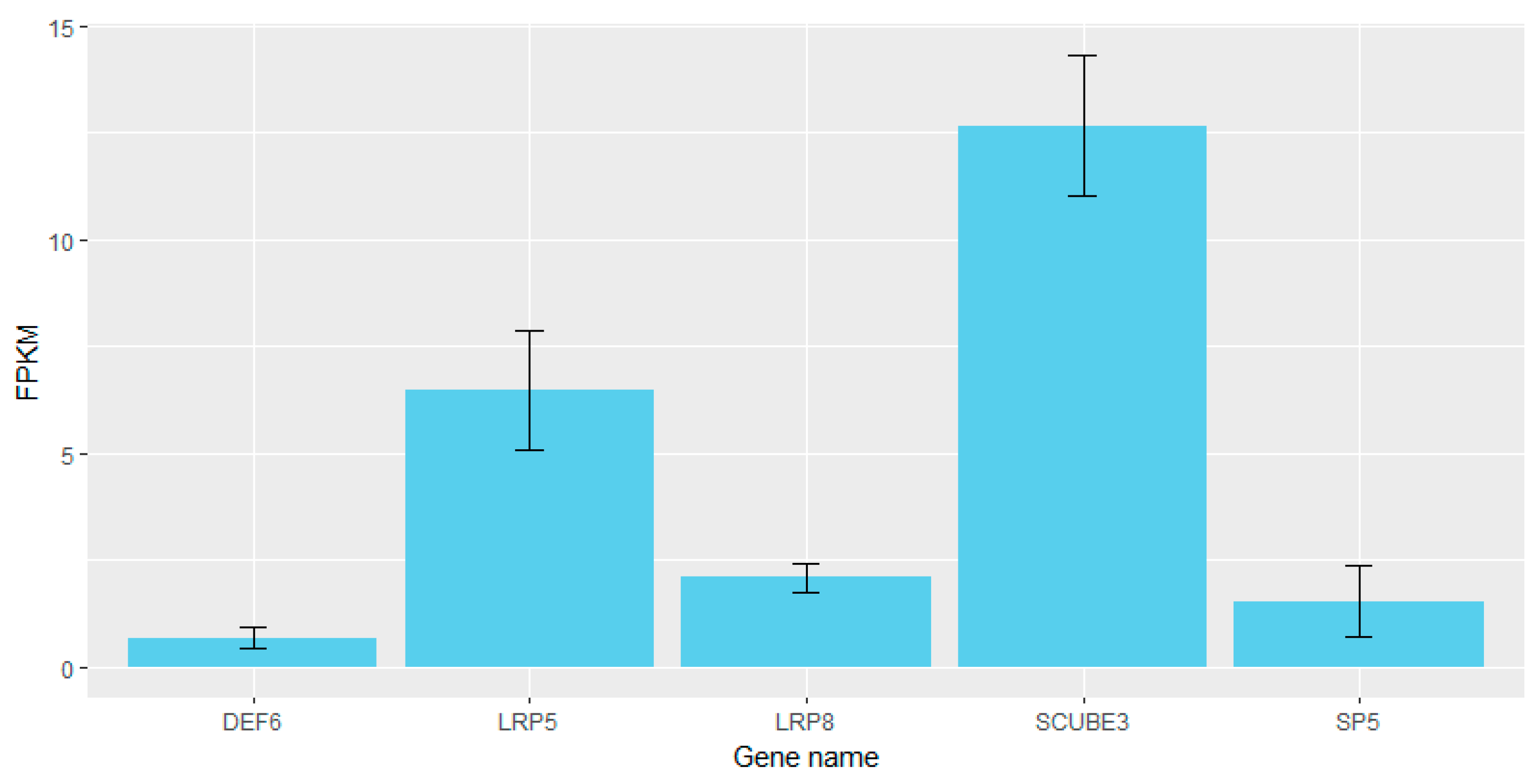
| 2 | Bone mineral content | 0 | 13,341,832 | LOC110259708, BEST1, FTH1, RAB3IL1, FADS3, INCENP, C2H11orf24, CHKA, KMT5B, LOC102162815, LOC100738812, LRP5, LOC110259247, FADS2, LOC106509334 |
| 2 | Lumbar vertebra number | 21,047,891 | 146,185,081 | LOC106509513, LOC102164448, PAM, YTHDC2, SLCO4C1, LOC110255325, LOC102158973, LOC110259457, PPIC, SNX24, C2H11orf91, CD59, FBXO3, KIAA1549L, LOC106509385 |
| 6 | Thoracic vertebra number | 158,835,021 | 158,835,025 | LRP8 2 |
| 7 | Cervical vertebra length | 28,939,911 | 38,532,223 | DEF6, TCP11, ZNF76, SCUBE3, PPARD |
| 7 | Cannon bone circumference | 31,235,547 | 31,235,551 | PPARD |
| 7 | Number of ribs | 68,061,952 | 77,142,053 | LOC100736765, MYH7, NGDN, ZFHX2 |
| 7 | Thoracic vertebra number | 104,557,779 | 104,557,783 | FOXN3 2 |
| 8 | Spinal curvature | 6,099,724 | 6,099,728 | LYAR |
| 10 | Vertebra number | 13,546,750 | 69,196,799 | CELF2, LOC110255590, LOC106505197, USP6NL, LOC110255605, LOC110255591, ARMC3, LOC106505172 |
| 12 | Thoracic vertebra number | 0 | 50,491,372 | BPTF, C12H17orf58, LOC110256159, KPNA2, TRNAR-CCG, HELZ, CA10 |
| 13 | Vertebra number | 75,687,809 | 169,726,013 | CMSS1, FILIP1L, LOC100524713 |
| 14 | Cervical vertebra length | 62,245,247 | 81,061,212 | LOC110256649, LOC110256651, PHYHIPL, FAM13C |
| 16 | Number of ribs | 28,477,997 | 34,396,617 | LOC110257315, ARL15, NDUFS4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Wang, M.; He, X.; Dong, R.; Liu, H.; Yu, G. Detection and Analysis of Genes Affecting the Number of Thoracic Vertebrae in Licha Black Pigs. Genes 2024, 15, 477. https://doi.org/10.3390/genes15040477
Wang Y, Wang M, He X, Dong R, Liu H, Yu G. Detection and Analysis of Genes Affecting the Number of Thoracic Vertebrae in Licha Black Pigs. Genes. 2024; 15(4):477. https://doi.org/10.3390/genes15040477
Chicago/Turabian StyleWang, Yuan, Min Wang, Xiaojin He, Ruilan Dong, Hongjiang Liu, and Guanghui Yu. 2024. "Detection and Analysis of Genes Affecting the Number of Thoracic Vertebrae in Licha Black Pigs" Genes 15, no. 4: 477. https://doi.org/10.3390/genes15040477
APA StyleWang, Y., Wang, M., He, X., Dong, R., Liu, H., & Yu, G. (2024). Detection and Analysis of Genes Affecting the Number of Thoracic Vertebrae in Licha Black Pigs. Genes, 15(4), 477. https://doi.org/10.3390/genes15040477