
Spectrum of ERCC6-Related Cockayne Syndrome (Type B): From Mild to Severe Forms

 , , , , , , , , and
, , , , , , , , and 
Abstract
1. Introduction
2. Materials and Methods
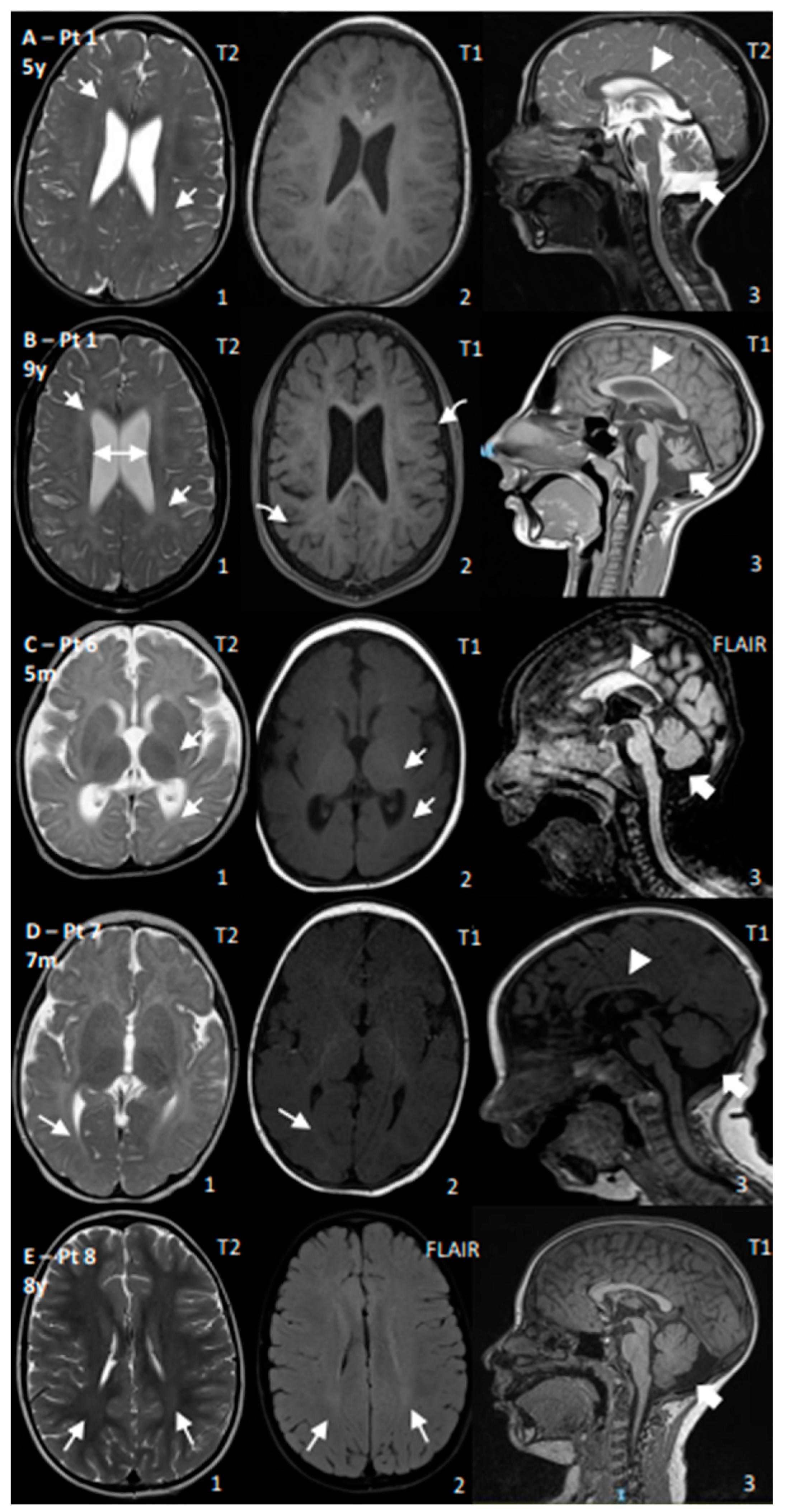
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laugel, V. Cockayne syndrome: The expanding clinical and mutational spectrum. Mech. Ageing Dev. 2013, 134, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Stafki, S.A.; Turner, J.; Littel, H.R.; Bruels, C.C.; Truong, D.; Knirsch, U.; Stettner, G.M.; Graf, U.; Berger, W.; Kinali, M.; et al. The Spectrum of MORC2-Related Disorders: A Potential Link to Cockayne Syndrome. Pediatr. Neurol. 2023, 141, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Laugel, V.; Dalloz, C.; Tobias, E.S.; Tolmie, J.L.; Martin-Coignard, D.; Drouin-Garraud, V.; Valayannopoulos, V.; Sarasin, A.; Dollfus, H. Cerebro-oculo-facio-skeletal syndrome: Three additional cases with CSB mutations, new diagnostic criteria and an approach to investigation. J. Med. Genet. 2008, 45, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, E.; Mustonen, A.; Olsen, P.; Miettinen, S.; Savuoja, T.; Raams, A.; Jaspers, N.G.J.; Shao, H.; Wu, B.L.; Ignatius, J. ERCC6 founder mutation identified in Finnish patients with COFS syndrome. Clin. Genet. 2010, 78, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Karikkineth, A.C.; Scheibye-Knudsen, M.; Fivenson, E.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res. Rev. 2017, 33, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Nance, M.A.; Berry, S.A.; Berry, S.A. Cockayne syndrome: Review of 140 cases. Am. J. Med. Genet. 1992, 42, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Spitz, M.A.; Severac, F.; Obringer, C.; Baer, S.; Le May, N.; Calmels, N.; Laugel, V. Diagnostic and severity scores for Cockayne syndrome. Orphanet J. Rare Dis. 2021, 16, 63. [Google Scholar] [CrossRef]
- Laugel, V.; Dalloz, C.; Durand, M.; Sauvanaud, F.; Kristensen, U.; Vincent, M.C.; Pasquier, L.; Odent, S.; Cormier-Daire, V.; Gener, B.; et al. Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum. Mutat. 2010, 31, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Chikhaoui, A.; Kraoua, I.; Calmels, N.; Bouchoucha, S.; Obringer, C.; Zayoud, K.; Montagne, B.; M’rad, R.; Abdelhak, S.; Laugel, V.; et al. Heterogeneous clinical features in Cockayne syndrome patients and siblings carrying the same CSA mutations. Orphanet J. Rare Dis. 2022, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Baer, S.; Tuzin, N.; Kang, P.B.; Mohammed, S.; Kubota, M.; van Ierland, Y.; Busa, T.; Rossi, M.; Morel, G.; Michot, C.; et al. Growth charts in Cockayne syndrome type 1 and type 2. Eur. J. Med. Genet. 2021, 64, 104105. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.T.; Stark, Z.; Sutton, R.E.; Danda, S.; Ekbote, A.V.; Elsayed, S.M.; Gibson, L.; Goodship, J.A.; Jackson, A.P.; Keng, W.T.; et al. The Cockayne Syndrome Natural History (CoSyNH) study: Clinical findings in 102 individuals and recommendations for care. Genet. Med. 2016, 18, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Duong, N.T.; Anh, N.P.; Bac, N.D.; Quang, L.B.; Miyake, N.; Van Hai, N.; Matsumoto, N. Whole-exome sequencing revealed a novel ERCC6 variant in a Vietnamese patient with Cockayne syndrome. Hum. Genome Var. 2022, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Gültekin-Zaim, Ö.B.; Yalçın-Çakmaklı, G.; Çolpak, A.İ.; Şimşek-Kiper, P.Ö.; Utine, G.E.; Elibol, B. Cockayne syndrome type 3 with dystonia-ataxia and clicking blinks. Mov. Disord. Clin. Pract. 2023, 10, S48–S50. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Mishima, T.; Fujioka, S.; Izumi, K.; Ando, M.; Higuchi, Y.; Takashima, H.; Tsuboi, Y. Siblings with Cockayne Syndrome B Type III Presenting with Slowly Progressive Cerebellar Ataxia. Intern. Med. 2023, 62, 2253–2259. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, M.; Nakano, E.; Nakazawa, Y.; Kanda, F.; Ueda, T.; Ogi, T.; Nishigori, C. A case of Cockayne syndrome with unusually mild clinical manifestations. J. Dermatol. 2023, 50, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Zhao, A.; Wang, Y.; Cao, Q.; Pan, C.; Li, M. Next-generation sequencing through multi-gene panel testing for the diagnosis of a Chinese patient with atypical Cockayne syndrome. Mol. Genet. Genom. Med. 2023, 11, e2254. [Google Scholar] [CrossRef]
- Cordts, I.; Önder, D.; Traschütz, A.; Kobeleva, X.; Karin, I.; Minnerop, M.; Koertvelyessy, P.; Biskup, S.; Forchhammer, S.; Binder, J.; et al. Adult-Onset Neurodegeneration in Nucleotide Excision Repair Disorders (NERDND): Time to Move beyond the Skin. Mov. Disord. 2022, 37, 1707–1718. [Google Scholar] [CrossRef]
- Koob, M.; Laugel, V.; Durand, M.; Fothergill, H.; Dalloz, C.; Sauvanaud, F.; Dollfus, H.; Namer, I.J.; Dietemann, J.L. Neuroimaging in Cockayne syndrome. Am. J. Neuroradiol. 2010, 31, 1623–1630. [Google Scholar] [CrossRef]
- Troelstra, C.; van Gool, A.; de Wit, J.; Vermeulen, W.; Bootsma, D.; Hoeijmakers, J.H.J. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell 1992, 71, 939–953. [Google Scholar] [CrossRef]
- Aamann, M.D.; Muftuoglu, M.; Bohr, V.A.; Stevnsner, T. Multiple interaction partners for Cockayne syndrome proteins: Implications for genome and transcriptome maintenance. Mech. Ageing Dev. 2013, 134, 212–224. [Google Scholar] [CrossRef]
- Berquist, B.R.; Canugovi, C.; Sykora, P.; Wilson, D.M.; Bohr, V.A. Human Cockayne syndrome B protein reciprocally communicates with mitochondrial proteins and promotes transcriptional elongation. Nucleic Acids Res. 2012, 40, 8392–8405. [Google Scholar] [CrossRef] [PubMed]
- Ghai, S.J.; Shago, M.; Shroff, M.; Yoon, G. Cockayne syndrome caused by paternally inherited 5 Mb deletion of 10q11.2 and a frameshift mutation of ERCC6. Eur. J. Med. Genet. 2011, 54, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Millere, E.; Rots, D.; Simrén, J.; Ashton, N.J.; Kupats, E.; Micule, I.; Priedite, V.; Kurjane, N.; Blennow, K.; Gailite, L.; et al. Plasma neurofilament light chain as a potential biomarker in Charcot-Marie-Tooth disease. Eur. J. Neurol. 2021, 28, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Beerepoot, S.; Heijst, H.; Roos, B.; Wamelink, M.M.C.; Boelens, J.J.; Lindemans, C.A.; Van Hasselt, P.M.; Jacobs, E.H.; Van Der Knaap, M.S.; Teunissen, C.E.; et al. Neurofilament light chain and glial fibrillary acidic protein levels in metachromatic leukodystrophy. Brain 2022, 145, 105. [Google Scholar] [CrossRef] [PubMed]
- Kessler, C.; Serna-Higuita, L.M.; Rattay, T.W.; Maetzler, W.; Wurster, I.; Hayer, S.; Wilke, C.; Hengel, H.; Reichbauer, J.; Armbruster, M.; et al. Neurofilament light chain is a cerebrospinal fluid biomarker in hereditary spastic paraplegia. Ann. Clin. Transl. Neurol. 2021, 8, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Meier, S.; Schaedelin, S.; Manouchehrinia, A.; Yaldizli, Ö.; Maceski, A.; Oechtering, J.; Achtnichts, L.; Conen, D.; Derfuss, T.; et al. Serum neurofilament light chain for individual prognostication of disease activity in people with multiple sclerosis: A retrospective modelling and validation study. Lancet Neurol. 2022, 21, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J. Blinded by the UV light: How the focus on transcription-coupled NER has distracted from understanding the mechanisms of Cockayne syndrome neurologic disease. DNA Repair 2013, 12, 656–671. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.F.C.; Bozek, K.; Herholz, M.; Trifunovic, A.; Rieckher, M.; Schumacher, B. A C. elegans model for neurodegeneration in Cockayne syndrome. Nucleic Acids Res. 2020, 48, 10973–10985. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Patient | CS Type | Age (Years) | Sex | Age at Diagnosis (Years) | Pathogenic Variants | Genetic Classification |
|---|---|---|---|---|---|---|
| 1 | CS I | 17 | F | 17 | ERCC6: homozygous c.2203C>T; p.Arg735Ter | CSB |
| 2 | CS I | 7 | F | 3 | ERCC6: c.2203C>T, p.Arg735Ter; 10q11.22-q11.23 microdeletion | CSB |
| 3 | CS I | 16 | M | 4.5 | ERCC6: c. 2203C>T, p.Arg735Ter; c.2096_2097insC, p.Leu700ValfsTer60 | CSB |
| 4 | CS I | 21 | M | 5 | ERCC6: c.2751C>T, p.Gly917Gly paternally inherited; c.2203C>T, p.Arg735Ter paternally inherited; chromosome 10 5.5Mb deletion maternally inherited | CSB |
| 5 | CS II | 2.5 | M | 1 | ERCC6: c.2784_2785delAG, p.Arg 928fsTer5; c.1881C>A, p.Tyr627Ter | CSB |
| 6 | CS II | 5 | F | 1 | ERCC6: homozygous c.1431_1432delGA, p.K478TfsTer9 | CSB |
| 7 | CS II | 3 | M | 1 | ERCC6: c.2383-1G>T, splice acceptor variant; c.3259C>T, p.Arg1087Ter | CSB |
| 8 | CS III | 9 | M | 9 | ERCC6: c.2096dupC, p.Leu700fsTer60; c.2291T>C, p.Leu764Ser | CSB |
| Patient CS Type | Ophtalmologic | Gestation and Birth | Photosensitivity | Growth Failure | Dysphagia | Hypertransaminasemia | Other Features | Diagnostic Score: CS; CRS | Severity Score |
|---|---|---|---|---|---|---|---|---|---|
| 1 CS I | Normal vision | Normal | Yes | Yes, <−5 SD | No | Yes (at age 1 y), then normalized | - | 9/20; 19/39 | 5/15 |
| 2 CS I | Small opacity in left eye lens; reduced retinic pigmentation (salt and pepper appearance) | Reduced head circumference growth during gestation | Yes | Yes, −2 SD | Yes | Yes (at age 4 y), then normalized | - | 8/20; 16/39 | 8/15 |
| 3 CS I | Astigmatism | CSec at 37 GW due to fetal bradycardia | Yes | Yes, <−5 SD | Yes | Yes | Photophobia. Cryptorchdism. | 7/20; 13/39 | 4/15 |
| 4 CS I | Hypermetropy | Normal | Yes | Yes, <−5 SD | Yes | Yes | Scoliosis. Cryptorchidism. | 7/20; N/A | 3/15 |
| 5 CS II | Normal vision | Reduced growth during last month | Yes | Yes, <−5 SD | No | Yes | Enophtalmia. | 10/20; N/A | 2/15 |
| 6 CS II | Bilateral congenital cataract, with recurrence after surgical treatment | CSec at 36 GW due to growth restriction; SGA at birth | Yes | Yes, <−5 SD | Yes | Yes | - | 10/20; 16/39 | 1/15 |
| 7 CS II | Hypermetropy | Right clubfoot and bilateral pyelectasis | No | Yes, <−5 SD | Yes | Yes | Thoracic kiphosis. Cryptorchidism. Enophtalmia. | 8/20; 13/39 | 2/15 |
| 8 CS III | Normal vision | Normal | No | No | No | No | Monolateral kidney dysplasia. | 0/20; 3/39 | 14/15 |
| Pat. | CS Type | Spasticity | Ataxia | Seizures | Microcephaly | Maximum Developmental Milestone (Age) | Language Development | sNFL (pg/mL) | Brain MRI Findings | Neurophisiologic Findings |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CS I | Yes | Yes | No | Yes, −4 SD | Walk (2 y, lost 3 y) | Poor | 45 | Age: 4 y and 9 y: Progressive cerebral and cerebellar atrophy; thin CC; permanent hypomyelination | ERG: normal; VEP: increased latency; BAEP: increased I–V latency; SSEP: increased CCT |
| 2 | CS I | Yes | Yes | No | Yes, −3 SD | Walk with support (2 y) | Poor | 99–136 | Age: 10 m and 1.5 y: Progressive cerebral and cerebellar atrophy; thin CC; WM reduction and hypomyelination | NCV: normal |
| 3 | CS I | Yes | Yes | No | Yes, <−5 SD | Walk with support (1.7 y) | Poor | N/A | Age: 4 y: Permanent hypomyelination | ERG: reduced amplitude |
| 4 | CS I | Yes | No | No | Yes, <−5 SD | Walk (1 y), then regression | Poor | N/A | N/A | N/A |
| 5 | CS II | Yes | No | No | Yes, <−5 SD | Head control (4.5 m) | Poor | 175 | N/A | N/A |
| 6 | CS II | Yes | Yes | Yes | Yes, <−5 SD | No acquisition | No acquisition | N/A | Age: 5 m: Cerebral, cerebellar, CC, and WM reductions; reduced myelination for age | EEG: occipital anomalies; VEP: poor cortical definition; BAEP: increased I–V latency |
| 7 | CS II | Yes | Yes | No | Yes, <−5 SD | Head control (5 m) | Poor, then regression | 198–270 | Age 7 m: Cerebral, cerebellar, CC, and WM reductions; reduced myelination for age | NCV: demyelinating SM neuropathy; ERG: normal; VEP: high latency |
| 8 | CS III | No | Yes | No | No | Walk (1 y) | Normal | 49 | Age: 8 y: mild posterior WM T2 hyperintensity; increased posterior fossa | BAEPs: increased I–V latency; ERG/VEP: normal; NCS: mild decrease in motor conduction velocity in the lower limbs |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sartorelli, J.; Travaglini, L.; Macchiaiolo, M.; Garone, G.; Gonfiantini, M.V.; Vecchio, D.; Sinibaldi, L.; Frascarelli, F.; Ceccatelli, V.; Petrillo, S.; et al. Spectrum of ERCC6-Related Cockayne Syndrome (Type B): From Mild to Severe Forms. Genes 2024, 15, 508. https://doi.org/10.3390/genes15040508
Sartorelli J, Travaglini L, Macchiaiolo M, Garone G, Gonfiantini MV, Vecchio D, Sinibaldi L, Frascarelli F, Ceccatelli V, Petrillo S, et al. Spectrum of ERCC6-Related Cockayne Syndrome (Type B): From Mild to Severe Forms. Genes. 2024; 15(4):508. https://doi.org/10.3390/genes15040508
Chicago/Turabian StyleSartorelli, Jacopo, Lorena Travaglini, Marina Macchiaiolo, Giacomo Garone, Michaela Veronika Gonfiantini, Davide Vecchio, Lorenzo Sinibaldi, Flaminia Frascarelli, Viola Ceccatelli, Sara Petrillo, and et al. 2024. "Spectrum of ERCC6-Related Cockayne Syndrome (Type B): From Mild to Severe Forms" Genes 15, no. 4: 508. https://doi.org/10.3390/genes15040508
APA StyleSartorelli, J., Travaglini, L., Macchiaiolo, M., Garone, G., Gonfiantini, M. V., Vecchio, D., Sinibaldi, L., Frascarelli, F., Ceccatelli, V., Petrillo, S., Piemonte, F., Piccolo, G., Novelli, A., Longo, D., Pro, S., D’Amico, A., Bertini, E. S., & Nicita, F. (2024). Spectrum of ERCC6-Related Cockayne Syndrome (Type B): From Mild to Severe Forms. Genes, 15(4), 508. https://doi.org/10.3390/genes15040508