
Research Progress and Applications of Bovine Genome in the Tribe Bovini

Abstract
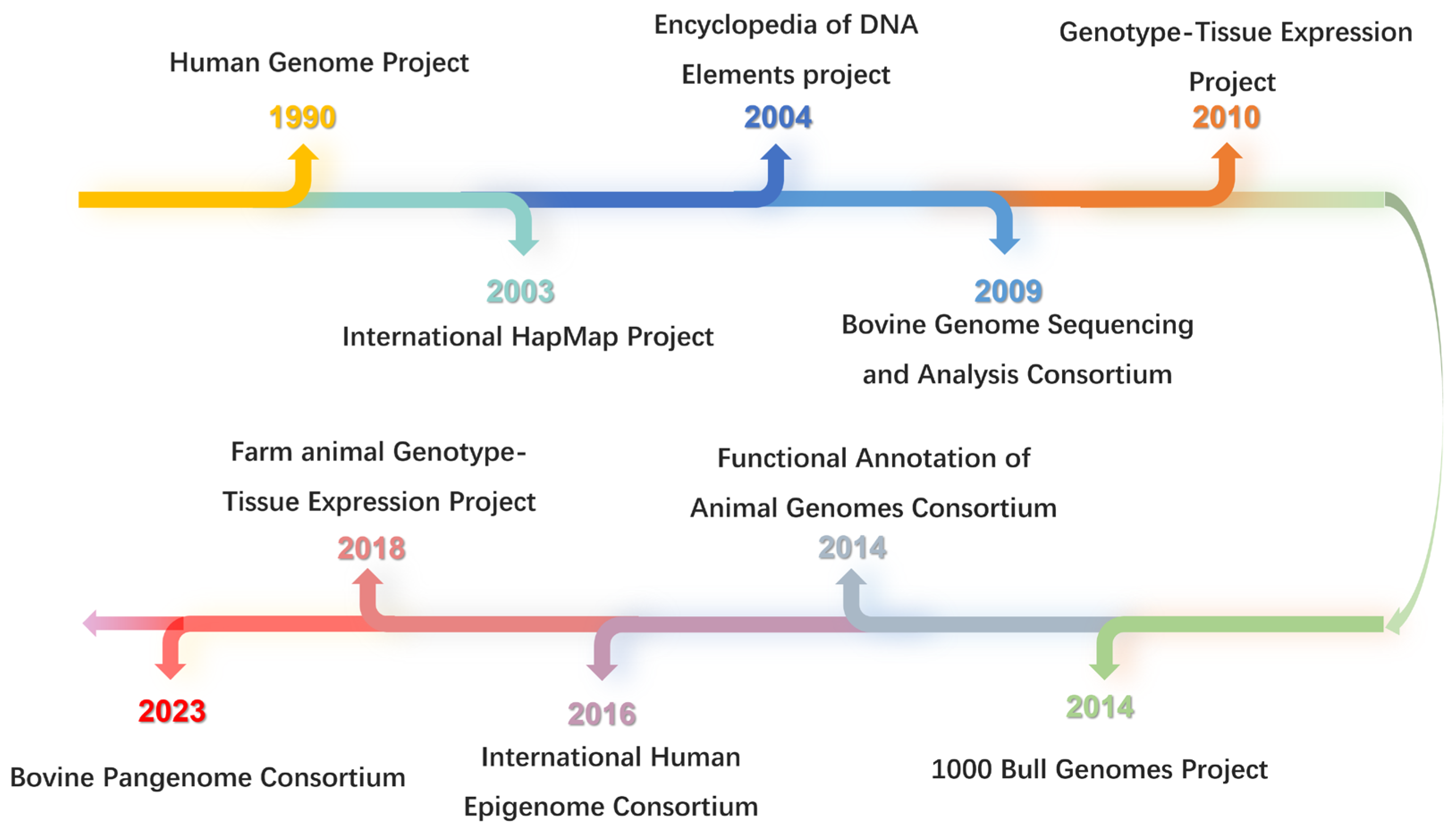
1. Introduction
2. Development of the Tribe Bovini Genome
3. Bovine Pangenome Studies
4. Comprehensive Functional Annotation of the Bovine Genome
5. Research Focus and Applications of the Bovine Genome
6. Advances in the Adaptive Evolution of Bovine
7. Construction and Application of Bovine Genome Datasets
8. Challenges in the Bovine Genome and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Ding, X.Z.; Liang, C.N.; Guo, X.; Wu, X.Y.; Wang, H.B.; Johnson, K.A.; Yan, P. Physiological insight into the high-altitude adaptations in domesticated yaks (Bos grunniens) along the Qinghai-Tibetan Plateau altitudinal gradient. Livest. Sci. 2014, 162, 233–239. [Google Scholar] [CrossRef]
- Ayalew, W.; Chu, M.; Liang, C.; Wu, X.; Yan, P. Adaptation Mechanisms of Yak (Bos grunniens) to High-Altitude Environmental Stress. Animals 2021, 11, 2344. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Long, R.; Ding, Y.; Wang, J.; Ding, L.; Wang, H. Morphological adaptations of yak (Bos grunniens) tongue to the foraging environment of the Qinghai-Tibetan Plateau1. J. Anim. Sci. 2010, 88, 2594–2603. [Google Scholar] [CrossRef]
- Liu, X.; Gao, J.; Liu, S.; Cheng, Y.; Hao, L.; Liu, S.; Zhu, W. The uniqueness and superiority of energy utilization in yaks compared with cattle in the highlands: A review. Anim. Nutr. 2023, 12, 138–144. [Google Scholar] [CrossRef]
- Kumar, S.; Nagarajan, M.; Sandhu, J.S.; Kumar, N.; Behl, V.; Nishanth, G. Mitochondrial DNA analyses of Indian water buffalo support a distinct genetic origin of river and swamp buffalo. Anim. Genet. 2007, 38, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Bickhart, D.M.; Ramunno, L.; Iamartino, D.; Williams, J.L.; Liu, G.E. Comparative sequence alignment reveals River Buffalo genomic structural differences compared with cattle. Genomics 2019, 111, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.W. Genetic and Biological Aspects of Zebu Adaptability. J. Anim. Sci. 1980, 50, 1201–1205. [Google Scholar] [CrossRef]
- Uzzaman, M.d.R.; Bhuiyan, M.d.S.A.; Edea, Z.; Kim, K.-S. Semi-domesticated and Irreplaceable Genetic Resource Gayal (Bos frontalis) Needs Effective Genetic Conservation in Bangladesh: A Review. Asian Australas. J. Anim. Sci. 2014, 27, 1368–1372. [Google Scholar] [CrossRef]
- Watson, J.D. The human genome project: Past, present, and future. Science 1990, 248, 44–49. [Google Scholar] [CrossRef]
- International HapMap Consortium. The International HapMap Project. Nature 2003, 426, 789–796. [Google Scholar] [CrossRef]
- ENCODE Project Consortium; Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigó, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium Human Genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Elsik, C.G.; Tellam, R.L.; Worley, K.C.; Gibbs, R.A.; Muzny, D.M.; Weinstock, G.M.; Adelson, D.L.; Eichler, E.E.; Elnitski, L.; Guigó, R.; et al. The Genome Sequence of Taurine Cattle: A Window to Ruminant Biology and Evolution. Science 2009, 324, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.A.; Taylor, J.F.; Van Tassell, C.P.; Barendse, W.; Eversole, K.A.; Gill, C.A.; Green, R.D.; Hamernik, D.L.; Kappes, S.M.; Lien, S.; et al. Genome-Wide Survey of SNP Variation Uncovers the Genetic Structure of Cattle Breeds. Science 2009, 324, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Daetwyler, H.D. 1000 Bull Genomes Project to Map Simple and Complex Genetic Traits in Cattle: Applications and Outcomes. Annu. Rev. Anim. Biosci. 2019, 7, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Fu, W.; Zhao, J.; Shen, J.; Chen, Q.; Zheng, Z.; Chen, H.; Sonstegard, T.S.; Lei, C.; Jiang, Y. BGVD: An Integrated Database for Bovine Sequencing Variations and Selective Signatures. Genom. Proteom. Bioinform. 2020, 18, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.P.L.; Bickhart, D.M.; Boichard, D.; Chamberlain, A.J.; Djikeng, A.; Jiang, Y.; Low, W.Y.; Pausch, H.; Demyda-Peyrás, S.; Prendergast, J.; et al. The Bovine Pangenome Consortium: Democratizing production and accessibility of genome assemblies for global cattle breeds and other bovine species. Genome Biol. 2023, 24, 139. [Google Scholar] [CrossRef]
- Giuffra, E.; Tuggle, C.K.; FAANG Consortium. Functional Annotation of Animal Genomes (FAANG): Current Achievements and Roadmap. Annu. Rev. Anim. Biosci. 2019, 7, 65–88. [Google Scholar] [CrossRef]
- Liu, S.; Gao, Y.; Canela-Xandri, O.; Wang, S.; Yu, Y.; Cai, W.; Li, B.; Xiang, R.; Chamberlain, A.J.; Pairo-Castineira, E.; et al. A multi-tissue atlas of regulatory variants in cattle. Nat. Genet. 2022, 54, 1438–1447. [Google Scholar] [CrossRef]
- Canavez, F.C.; Luche, D.D.; Stothard, P.; Leite, K.R.M.; Sousa-Canavez, J.M.; Plastow, G.; Meidanis, J.; Souza, M.A.; Feijao, P.; Moore, S.S.; et al. Genome Sequence and Assembly of Bos indicus. J. Hered. 2012, 103, 342–348. [Google Scholar] [CrossRef]
- Qiu, Q.; Zhang, G.; Ma, T.; Qian, W.; Wang, J.; Ye, Z.; Cao, C.; Hu, Q.; Kim, J.; Larkin, D.M.; et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 2012, 44, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, W.; Liu, X.; Du, X.; Zhang, K.; Zhang, Y.; Song, Y.; Zi, Y.; Qiu, Q.; Lenstra, J.A.; et al. Structural Variants Selected during Yak Domestication Inferred from Long-Read Whole-Genome Sequencing. Mol. Biol. Evol. 2021, 38, 3676–3680. [Google Scholar] [CrossRef] [PubMed]
- Low, W.Y.; Tearle, R.; Bickhart, D.M.; Rosen, B.D.; Kingan, S.B.; Swale, T.; Thibaud-Nissen, F.; Murphy, T.D.; Young, R.; Lefevre, L.; et al. Chromosome-level assembly of the water buffalo genome surpasses human and goat genomes in sequence contiguity. Nat. Commun. 2019, 10, 260. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, T.; Xian, M.; Zhang, R.; Yang, W.; Su, B.; Yang, G.; Sun, L.; Xu, W.; Xu, S.; et al. A draft genome of Drung cattle reveals clues to its chromosomal fusion and environmental adaptation. Commun. Biol. 2022, 5, 353. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De novo assembly of the cattle reference genome with single-molecule sequencing. GigaScience 2020, 9, giaa021. [Google Scholar] [CrossRef] [PubMed]
- Heaton, M.P.; Smith, T.P.L.; Bickhart, D.M.; Vander Ley, B.L.; Kuehn, L.A.; Oppenheimer, J.; Shafer, W.R.; Schuetze, F.T.; Stroud, B.; McClure, J.C.; et al. A Reference Genome Assembly of Simmental Cattle, Bos taurus taurus. J. Hered. 2021, 112, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Eché, C.; Iampietro, C.; Birbes, C.; Dréau, A.; Kuchly, C.; Di Franco, A.; Klopp, C.; Faraut, T.; Djebali, S.; Castinel, A.; et al. A Bos taurus sequencing methods benchmark for assembly, haplotyping, and variant calling. Sci. Data 2023, 10, 369. [Google Scholar] [CrossRef]
- Gao, X.; Wang, S.; Wang, Y.-F.; Li, S.; Wu, S.-X.; Yan, R.-G.; Zhang, Y.-W.; Wan, R.-D.; He, Z.; Song, R.-D.; et al. Long read genome assemblies complemented by single cell RNA-sequencing reveal genetic and cellular mechanisms underlying the adaptive evolution of yak. Nat. Commun. 2022, 13, 4887. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, J.; Dou, J.; Yan, B.; Ren, Q.; Tang, B.; Wang, K.; Qiu, Q. The sequence and de novo assembly of the wild yak genome. Sci. Data 2020, 7, 66. [Google Scholar] [CrossRef]
- Williams, J.L.; Iamartino, D.; Pruitt, K.D.; Sonstegard, T.; Smith, T.P.L.; Low, W.Y.; Biagini, T.; Bomba, L.; Capomaccio, S.; Castiglioni, B.; et al. Genome assembly and transcriptome resource for river buffalo, Bubalus bubalis (2n = 50). GigaScience 2017, 6, gix088. [Google Scholar] [CrossRef]
- Mintoo, A.A.; Zhang, H.; Chen, C.; Moniruzzaman, M.; Deng, T.; Anam, M.; Emdadul Huque, Q.M.; Guang, X.; Wang, P.; Zhong, Z.; et al. Draft genome of the river water buffalo. Ecol. Evol. 2019, 9, 3378–3388. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zhou, Y.; Zhang, B.; Zhang, Y.; Wang, X.; Feng, T.; Li, Z.; Cui, K.; Wang, Z.; Luo, C.; et al. Understanding divergent domestication traits from the whole-genome sequencing of swamp- and river-buffalo populations. Natl. Sci. Rev. 2020, 7, 686–701. [Google Scholar] [CrossRef]
- Glanzmann, B.; Möller, M.; Le Roex, N.; Tromp, G.; Hoal, E.G.; Van Helden, P.D. The complete genome sequence of the African buffalo (Syncerus caffer). BMC Genom. 2016, 17, 1001. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-S.; Zeng, Y.; Wang, X.; Nie, W.-H.; Wang, J.-H.; Su, W.-T.; Otecko, N.O.; Xiong, Z.-J.; Wang, S.; Qu, K.-X.; et al. Draft genome of the gayal, Bos frontalis. GigaScience 2017, 6, gix094. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Cai, Z.; Mukherjee, A.; Longkumer, I.; Mech, M.; Vupru, K.; Khate, K.; Rajkhowa, C.; Mitra, A.; Guldbrandtsen, B.; et al. Whole genome sequence and de novo assembly revealed genomic architecture of Indian Mithun (Bos frontalis). BMC Genomics 2019, 20, 617. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, S.; Zhang, Z.; Luo, J.; Lin, G.L.; Deng, W.-D.; Guo, Z.; Han, F.M.; Wang, L.-L.; Li, J.; et al. Large-Scale Chromosomal Changes Lead to Genome-Level Expression Alterations, Environmental Adaptation, and Speciation in the Gayal (Bos frontalis). Mol. Biol. Evol. 2023, 40, msad006. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Bisht, M.S.; Saxena, R.; Mahajan, S.; Pulikkan, J.; Sharma, V.K. Genome sequencing and de novo and reference-based genome assemblies of Bos indicus breeds. Genes Genom. 2023, 45, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Rhie, A.; Walenz, B.P.; Dilthey, A.T.; Bickhart, D.M.; Kingan, S.B.; Hiendleder, S.; Williams, J.L.; Smith, T.P.L.; Phillippy, A.M. De novo assembly of haplotype-resolved genomes with trio binning. Nat. Biotechnol. 2018, 36, 1174–1182. [Google Scholar] [CrossRef]
- Crysnanto, D.; Leonard, A.S.; Fang, Z.-H.; Pausch, H. Novel functional sequences uncovered through a bovine multiassembly graph. Proc. Natl. Acad. Sci. USA 2021, 118, e2101056118. [Google Scholar] [CrossRef]
- Leonard, A.S.; Crysnanto, D.; Fang, Z.-H.; Heaton, M.P.; Vander Ley, B.L.; Herrera, C.; Bollwein, H.; Bickhart, D.M.; Kuhn, K.L.; Smith, T.P.L.; et al. Structural variant-based pangenome construction has low sensitivity to variability of haplotype-resolved bovine assemblies. Nat. Commun. 2022, 13, 3012. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, L.; Han, X.; Han, J.; Hu, Y.; Li, F.; Xia, H.; Peng, L.; Boschiero, C.; Rosen, B.D.; et al. Assembly of a pangenome for global cattle reveals missing sequences and novel structural variations, providing new insights into their diversity and evolutionary history. Genome Res. 2022, 32, 1585–1601. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Bian, P.; Hu, D.; Luo, F.; Huang, Y.; Jiao, S.; Wang, X.; Gong, M.; Li, R.; Cai, Y.; et al. A Chinese indicine pangenome reveals a wealth of novel structural variants introgressed from other Bos species. Genome Res. 2023, 33, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Harhay, G.P.; Smith, T.P.; Alexander, L.J.; Haudenschild, C.D.; Keele, J.W.; Matukumalli, L.K.; Schroeder, S.G.; Van Tassell, C.P.; Gresham, C.R.; Bridges, S.M.; et al. An atlas of bovine gene expression reveals novel distinctive tissue characteristics and evidence for improving genome annotation. Genome Biol. 2010, 11, R102. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, T.; Niu, Q.; Xu, L.; Chen, Y.; Gao, X.; Gao, H.; Zhang, L.; Liu, G.E.; Li, J.; et al. Transcriptional atlas analysis from multiple tissues reveals the expression specificity patterns in beef cattle. BMC Biol. 2022, 20, 79. [Google Scholar] [CrossRef] [PubMed]
- Goszczynski, D.E.; Halstead, M.M.; Islas-Trejo, A.D.; Zhou, H.; Ross, P.J. Transcription initiation mapping in 31 bovine tissues reveals complex promoter activity, pervasive transcription, and tissue-specific promoter usage. Genome Res. 2021, 31, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Liu, S.; Liu, M.; Kang, X.; Lin, S.; Li, B.; Connor, E.E.; Baldwin, R.L.; Tenesa, A.; Ma, L.; et al. Functional annotation of the cattle genome through systematic discovery and characterization of chromatin states and butyrate-induced variations. BMC Biol. 2019, 17, 68. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, P.A.; Naval-Sánchez, M.; Menzies, M.; Nguyen, L.T.; Porto-Neto, L.R.; Fortes, M.R.S.; Reverter, A. Chromatin accessibility and regulatory vocabulary across indicine cattle tissues. Genome Biol. 2021, 22, 273. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liu, S.; Shi, S.; Jiang, Y.; Cao, M.; Tang, Y.; Li, W.; Liu, J.; Fang, L.; Yu, Y.; et al. Comparative epigenomics reveals the impact of ruminant-specific regulatory elements on complex traits. BMC Biol. 2022, 20, 273. [Google Scholar] [CrossRef] [PubMed]
- Daetwyler, H.D.; Capitan, A.; Pausch, H.; Stothard, P.; Van Binsbergen, R.; Brøndum, R.F.; Liao, X.; Djari, A.; Rodriguez, S.C.; Grohs, C.; et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 2014, 46, 858–865. [Google Scholar] [CrossRef]
- Bouwman, A.C.; Daetwyler, H.D.; Chamberlain, A.J.; Ponce, C.H.; Sargolzaei, M.; Schenkel, F.S.; Sahana, G.; Govignon-Gion, A.; Boitard, S.; Dolezal, M.; et al. Meta-analysis of genome-wide association studies for cattle stature identifies common genes that regulate body size in mammals. Nat. Genet. 2018, 50, 362–367. [Google Scholar] [CrossRef]
- Qiu, Q.; Wang, L.; Wang, K.; Yang, Y.; Ma, T.; Wang, Z.; Zhang, X.; Ni, Z.; Hou, F.; Long, R.; et al. Yak whole-genome resequencing reveals domestication signatures and prehistoric population expansions. Nat. Commun. 2015, 6, 10283. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Luo, J.; Wang, F.; Xu, D.; Ahmed, Z.; Chen, S.; Li, R.; Ma, Z. Whole-genome resequencing reveals genetic diversity, differentiation, and selection signatures of yak breeds/populations in Qinghai, China. Front. Genet. 2023, 13, 1034094. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Nadeem, A.; Javed, M.; Hassan, F.; Luo, X.; Khalid, R.B.; Liu, Q. Genomic Identification, Evolution and Sequence Analysis of the Heat-Shock Protein Gene Family in Buffalo. Genes 2020, 11, 1388. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Hooda, O.K.; Singh, G.; Meur, S.K. Influence of induced heat stress on HSP70 in buffalo lymphocytes: HSP70 and buffalo lymphocytes. J. Anim. Physiol. Anim. Nutr. 2011, 95, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Surya, T.; Vineeth, M.R.; Sivalingam, J.; Tantia, M.S.; Dixit, S.P.; Niranjan, S.K.; Gupta, I.D. Genomewide identification and annotation of SNPs in Bubalus bubalis. Genomics 2019, 111, 1695–1698. [Google Scholar] [CrossRef] [PubMed]
- El-Bayomi, K.M.; Saleh, A.A.; Awad, A.; El-Tarabany, M.S.; El-Qaliouby, H.S.; Afifi, M.; El-Komy, S.; Essawi, W.M.; Almadaly, E.A.; El-Magd, M.A. Association of CYP19A1 gene polymorphisms with anoestrus in water buffaloes. Reprod. Fertil. Dev. 2018, 30, 487. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Deng, T.; Zhou, Y.; Ye, T.; Zhou, Z.; Zhang, S.; Shao, B.; Wei, P.; Sun, H.; Khan, F.A.; et al. Systematic analyses for candidate genes of milk production traits in water buffalo (Bubalus bubalis). Anim. Genet. 2019, 50, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Liang, A.X.; Campanile, G.; Plastow, G.; Zhang, C.; Wang, Z.; Salzano, A.; Gasparrini, B.; Cassandro, M.; Yang, L.G. Genome-wide association studies to identify quantitative trait loci affecting milk production traits in water buffalo. J. Dairy Sci. 2018, 101, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Cosenza, G.; Gaspa, G.; Iannaccone, M.; Macciotta, N.P.P.; Chemello, G.; Di Stasio, L.; Pauciullo, A. Sequencing of lipoprotein lipase gene in the Mediterranean river buffalo identified novel variants affecting gene expression. J. Dairy Sci. 2020, 103, 6374–6382. [Google Scholar] [CrossRef]
- Liang, D.; Zhao, P.; Si, J.; Fang, L.; Pairo-Castineira, E.; Hu, X.; Xu, Q.; Hou, Y.; Gong, Y.; Liang, Z.; et al. Corrigendum to: Genomic analysis revealed a convergent evolution of LINE-1 in coat color: A case study in water buffaloes (B. bubalis). Mol. Biol. Evol. 2021, 38, 2673. [Google Scholar] [CrossRef]
- Miao, Y.; Wu, G.; Wang, L.; Li, D.; Tang, S.; Liang, J.; Mao, H.; Luo, H.; Zhang, Y. The role of MC1R gene in buffalo coat color. Sci. China Life Sci. 2010, 53, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Iannaccone, M.; Cosenza, G.; Pauciullo, A.; Martino, G.; Capparelli, R. The SNP g.4667G>A at 3′-UTR of IFNG gene is associated with susceptibility to bovine tuberculosis in Mediterranean water buffalo (Bubalus bubalis). Anim. Genet. 2018, 49, 496–497. [Google Scholar] [CrossRef] [PubMed]
- El-Halawany, N.; Shawky, A.-E.A.; Al-Tohamy, A.F.M.; Hegazy, L.; Abdel-Shafy, H.; Abdel-Latif, M.A.; Ghazi, Y.A.; Neuhoff, C.; Salilew-Wondim, D.; Schellander, K. Complement component 3: Characterization and association with mastitis resistance in Egyptian water buffalo and cattle. J. Genet. 2017, 96, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Talenti, A.; Young, R.; Jayaraman, S.; Callaby, R.; Jadhav, S.K.; Dhanikachalam, V.; Manikandan, M.; Biswa, B.B.; Low, W.Y.; et al. Whole genome analysis of water buffalo and global cattle breeds highlights convergent signatures of domestication. Nat. Commun. 2020, 11, 4739. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hanotte, O.; Mwai, O.A.; Dessie, T.; Bashir, S.; Diallo, B.; Agaba, M.; Kim, K.; Kwak, W.; Sung, S.; et al. The genome landscape of indigenous African cattle. Genome Biol. 2017, 18, 34. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Liu, X.; Yang, T.; Huang, Z.; Hanif, Q.; Asif, M.; Khan, Q.M.; Mansoor, S. Genomic variants identified from whole-genome resequencing of indicine cattle breeds from Pakistan. PLoS ONE 2019, 14, e0215065. [Google Scholar] [CrossRef] [PubMed]
- Hanotte, O.; Dessie, T.; Kemp, S. Time to Tap Africa’s Livestock Genomes. Science 2010, 328, 1640–1641. [Google Scholar] [CrossRef]
- Xiang, R.; Berg, I.V.D.; MacLeod, I.M.; Hayes, B.J.; Prowse-Wilkins, C.P.; Wang, M.; Bolormaa, S.; Liu, Z.; Rochfort, S.J.; Reich, C.M.; et al. Quantifying the contribution of sequence variants with regulatory and evolutionary significance to 34 bovine complex traits. Proc. Natl. Acad. Sci. USA 2019, 116, 19398–19408. [Google Scholar] [CrossRef] [PubMed]
- Loftus, R.T.; MacHugh, D.E.; Bradley, D.G.; Sharp, P.M.; Cunningham, P. Evidence for two independent domestications of cattle. Proc. Natl. Acad. Sci. USA 1994, 91, 2757–2761. [Google Scholar] [CrossRef]
- Xia, X.; Qu, K.; Wang, Y.; Sinding, M.-H.S.; Wang, F.; Hanif, Q.; Ahmed, Z.; Lenstra, J.A.; Han, J.; Lei, C.; et al. Global dispersal and adaptive evolution of domestic cattle: A genomic perspective. Stress Biol. 2023, 3, 8. [Google Scholar] [CrossRef]
- McTavish, E.J.; Decker, J.E.; Schnabel, R.D.; Taylor, J.F.; Hillis, D.M. New World cattle show ancestry from multiple independent domestication events. Proc. Natl. Acad. Sci. USA 2013, 110, E1398–E1406. [Google Scholar] [CrossRef] [PubMed]
- Park, S.D.E.; Magee, D.A.; McGettigan, P.A.; Teasdale, M.D.; Edwards, C.J.; Lohan, A.J.; Murphy, A.; Braud, M.; Donoghue, M.T.; Liu, Y.; et al. Genome sequencing of the extinct Eurasian wild aurochs, Bos primigenius, illuminates the phylogeography and evolution of cattle. Genome Biol. 2015, 16, 234. [Google Scholar] [CrossRef] [PubMed]
- Barbato, M.; Hailer, F.; Upadhyay, M.; Del Corvo, M.; Colli, L.; Negrini, R.; Kim, E.-S.; Crooijmans, R.P.M.A.; Sonstegard, T.; Ajmone-Marsan, P. Adaptive introgression from indicine cattle into white cattle breeds from Central Italy. Sci. Rep. 2020, 10, 1279. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Cai, Y.; Chen, Q.; Li, R.; Wang, K.; Huang, Y.; Hu, S.; Huang, S.; Zhang, H.; Zheng, Z.; et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 2018, 9, 2337. [Google Scholar] [CrossRef]
- Xia, X.; Zhang, F.; Li, S.; Luo, X.; Peng, L.; Dong, Z.; Pausch, H.; Leonard, A.S.; Crysnanto, D.; Wang, S.; et al. Structural variation and introgression from wild populations in East Asian cattle genomes confer adaptation to local environment. Genome Biol. 2023, 24, 211. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, C.; Xu, H.; Xia, X.; Luo, X.; Li, K.; Han, J.; Lei, C.; Chen, N.; Yue, X. Genomic analysis reveals a KIT-related chromosomal translocation associated with the white coat phenotype in yak. J. Anim. Breed. Genet. 2023, 140, 330–342. [Google Scholar] [CrossRef]
- Shamimuzzaman, M.; Le Tourneau, J.J.; Unni, D.R.; Diesh, C.M.; Triant, D.A.; Walsh, A.T.; Tayal, A.; Conant, G.C.; Hagen, D.E.; Elsik, C.G. Bovine Genome Database: New annotation tools for a new reference genome. Nucleic Acids Res. 2019, 78, gkz944. [Google Scholar] [CrossRef] [PubMed]
- Raz, R.; Roth, Z.; Gershoni, M. ExAgBov: A public database of annotated variations from hundreds of bovine whole-exome sequencing samples. Sci. Data 2022, 9, 469. [Google Scholar] [CrossRef]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the future: Meeting new challenges and providing updated services. Nucleic Acids Res. 2022, 50, D956–D961. [Google Scholar] [CrossRef]
- Triant, D.A.; Walsh, A.T.; Hartley, G.A.; Petry, B.; Stegemiller, M.R.; Nelson, B.M.; McKendrick, M.M.; Fuller, E.P.; Cockett, N.E.; Koltes, J.E.; et al. AgAnimalGenomes: Browsers for viewing and manually annotating farm animal genomes. Mamm. Genome 2023, 34, 418–436. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, H.; Dou, J.; Wang, Y.; Liao, Y.; Huang, X.; Tang, Z.; Xu, J.; Yin, D.; Zhu, S.; et al. IAnimal: A cross-species omics knowledgebase for animals. Nucleic Acids Res. 2023, 51, D1312–D1324. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Yao, R.; Li, L.; Xu, Y.; Lei, B.; Tang, G.; Liang, H.; Lei, Y.; Li, C.; Li, X.; et al. A database of animal metagenomes. Sci. Data 2022, 9, 312. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Species Category | Breed | Genomics Version | Sequence Size (Gb) | Contig N50 (Mb) | Scafford N50 (Mb) | References |
|---|---|---|---|---|---|---|
| Cattle | Hereford | UMD B. taurus 2.0 | 2.87 | 0.08 | 1.25 | [13] |
| Hereford | ARS-UCD1.2 | 2.72 | 25.9 | 103.31 | [25] | |
| Simmental | ARS_Simm1.0 | 2.86 | 70.8 | 102 | [26] | |
| Charolais | -- | 3.2 | 87 | 88 | [27] | |
| Yak | Domestic yak | BosGru_v2.0 | 2.65 | 0.02 | 1.41 | [21] |
| Domestic yak | BosGru3.0 | 2.83 | 44.72 | 114.39 | [22] | |
| Domestic yak | -- | 2.61 | 44.91 | 104.02 | [28] | |
| Wild yak | -- | 2.83 | 0.06 | 16.3 | [29] | |
| Wild yak | -- | 2.63 | 38.28 | 103.9 | [22] | |
| Buffalo | Mediterranean | UMD_CASPUR_WB_2.0 | 2.83 | 0.022 | 1.41 | [30] |
| Bengal buffalo | Bubbub1.0 | 2.77 | 0.025 | 6.96 | [31] | |
| Italian Mediterranean buffalo | UOA_WB_1 | 2.65 | 18.8 | 117.2 | [23] | |
| Fuzhong swamp buffalo | -- | 2.63 | 8.8 | 117.3 | [32] | |
| Murrah river buffalo | -- | 2.64 | 3.1 | 116.1 | [32] | |
| African buffalo | -- | 2.68 | 0.043 | 2.4 | [33] | |
| Zebu | Nelore cattle | B. indicus_1.0 | 2.67 | 0.03 | 106.31 | [20] |
| Gayal | Drung cattle | -- | 2.85 | 0.01 | 2.74 | [34] |
| Nagaland | NRC_Mithun_1 | 3 | 0.028 | 1 | [35] | |
| Drung cattle | Drung_v1.2 | 2.74 | 0.157 | 4.08 | [24] | |
| Drung cattle | -- | 2.57 | 27.2 | -- | [36] |
| Project | Samples | Results | References |
|---|---|---|---|
| Bovine Digital Gene Atlas | Three growth stages (fetal, juvenile, and adult) and three cattle cell lines | This digital gene expression profile investigates the relationship between gene expression, tissue, and gene function. | [43] |
| Bovine Gene Expression Atlas | 135 bovine tissues in adult beef cattle, covering 51 tissue types | This study identified 19,356 novel transcripts; and detected 2654 HKGs, 477 TSGs, and 237 hub genes. | [44] |
| A Promoter Activity Atlas | 31 bovine tissues | This study identified and characterized transcription start sites, and shortened the gap between genotype and phenotype. | [45] |
| Bovine Epigenomic Landscape | Rumen | This study established the first global map of regulatory elements (15 chromatin states), and demonstrated the correlation among nutritional elements, chromatin states, gene activities, and phenotype outcomes. | [46] |
| Open Chromatin Profile | Liver, muscle, and hypothalamus | This study predicted potential master regulatory elements in these three tissues, namely, HNF4, MEF2, and SOX factors, respectively. | [47] |
| A Ruminant-Specific Regulatory Element Profile | Liver | This study systematically characterized the dynamic functional landscapes, and identified a core set (n = 6359) of ruminant-specific REs. | [48] |
| Cattle Genotype-Tissue Expression Atlas | More than 100 tissues/cell types | This study described the transcriptomic landscape, and evaluated the tissue-sharing patterns of genetic regulatory variants. | [19] |
| Species Category | Trait | Candidate Genes | References |
|---|---|---|---|
| Cattle | immune | IFNAR2, IL23R, IL24, IL15, LEAP2 | [13] |
| reproduction and dietary habitats | TAS2R46, QRICH2, PRDM9, and HSPA1A | [40] | |
| milk production | DGAT1 | [49] | |
| curly coat | KRT27 | [49] | |
| body size | PLAG1, NCAPG–LCORL | [50] | |
| Yak | sensory perception | GPCR, PLXNB1 | [21,51] |
| hypoxia response | ADAM17, ARG2, ARNT, GATA1, MAFG, KLF5, HOXB5, SFTPC, SCGB3A2, EPAS1 | [21,28] | |
| nutrient metabolism | CAMK2B, GCNT3, HSD17B12, WHSC1, and GLUL | [21] | |
| behavior | MAGI2, GAD2, GRIK2, ADCYAP1R1, SCRIB | [22,51] | |
| immunity | NAFT | [22] | |
| reproduction | SMOC2, TTLL1, RHPN1, RHOD | [22,51] | |
| disease resistance | CDK2AP2, PLEC, and CYB5B | [4] | |
| heat stress | NFAT5, HSF1, and SLC25A48 | [4] | |
| milk quality | OPLAH and GRINA | [4] | |
| neurodevelopment | SUSD4, INSYN1, PPP1CA, MAGI2 | [22,52] | |
| meat quality | ZRANB1 | [52] | |
| Buffalo | brain development and cognition | TEAD1, OXTR, ADYC3 | [32] |
| fecundity | ESR1 | [32] | |
| milk production | METTL17, RNASE2, RNASE4 | [32] | |
| body size | IGF2BP2 | [32] | |
| Zebu | heat tolerance | HSPA4, SOD1, PRLR, WNT, VSMC | [65,66] |
| resistance to tick infestation | BOLA | [65] | |
| hypoxia | VEGF, HIF-1 | [66] | |
| Gayal | cardiovascular function | MYH, DHPR, ROCK, MLCK2, RYR2, TNNI3, ACTC1 | [24] |
| muscle traits | TTN, NEB, MYH1, MYH2, MYH4 | [36] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, X.; Sun, Y.; Fu, T.; Gao, T.; Zhang, T. Research Progress and Applications of Bovine Genome in the Tribe Bovini. Genes 2024, 15, 509. https://doi.org/10.3390/genes15040509
Du X, Sun Y, Fu T, Gao T, Zhang T. Research Progress and Applications of Bovine Genome in the Tribe Bovini. Genes. 2024; 15(4):509. https://doi.org/10.3390/genes15040509
Chicago/Turabian StyleDu, Xingjie, Yu Sun, Tong Fu, Tengyun Gao, and Tianliu Zhang. 2024. "Research Progress and Applications of Bovine Genome in the Tribe Bovini" Genes 15, no. 4: 509. https://doi.org/10.3390/genes15040509
APA StyleDu, X., Sun, Y., Fu, T., Gao, T., & Zhang, T. (2024). Research Progress and Applications of Bovine Genome in the Tribe Bovini. Genes, 15(4), 509. https://doi.org/10.3390/genes15040509