
Genome Sequencing in an Individual Presenting with 22q11.2 Deletion Syndrome and Juvenile Idiopathic Arthritis

 and
and
Abstract
1. Introduction
2. Patient and Methods
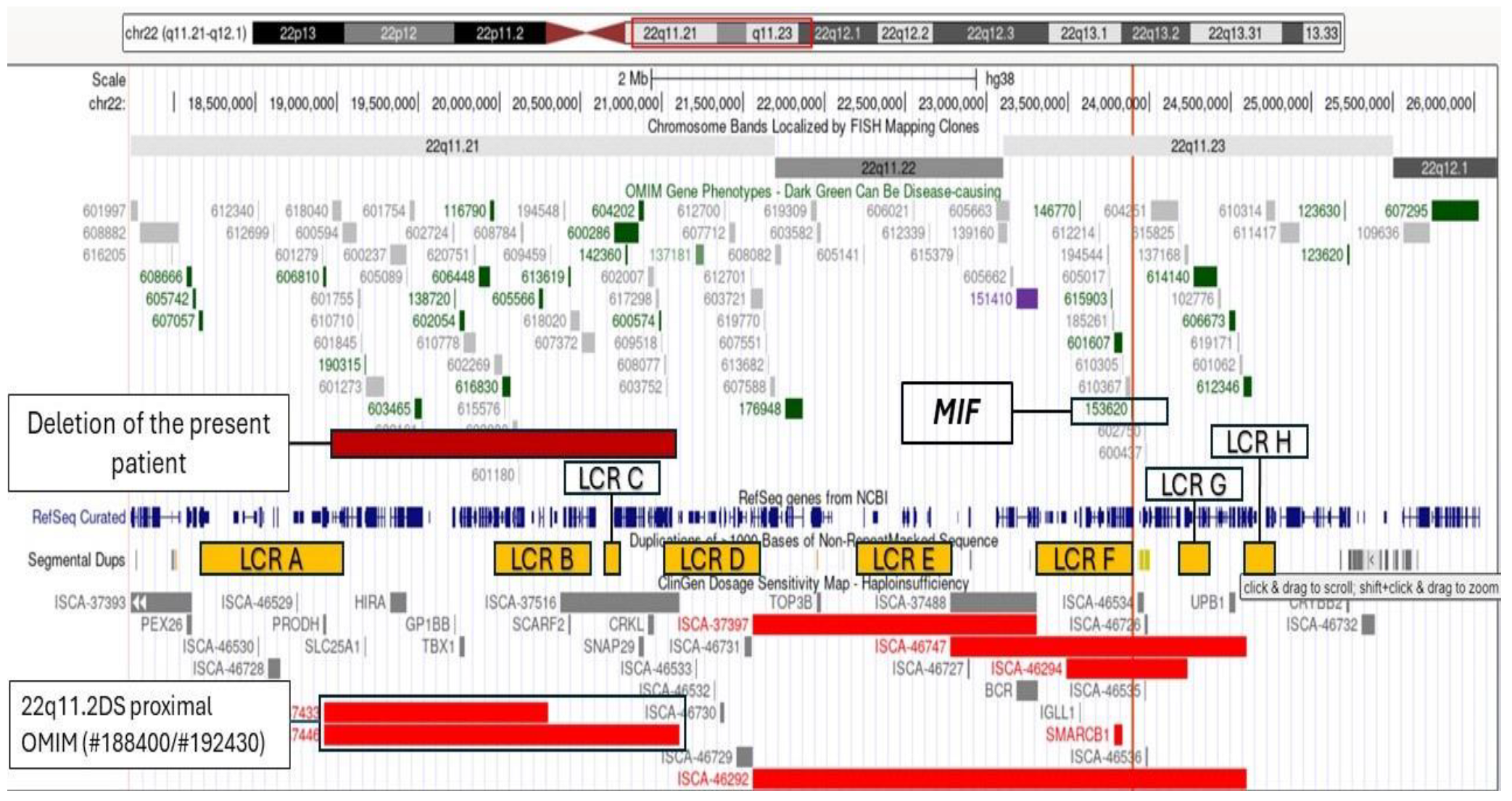
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Hou, X.; Qu, H.; Zhang, S.; Qi, X.; Hakonarson, H.; Xia, Q.; Li, J. The Multi-Omics Architecture of Juvenile Idiopathic Arthritis. Cells 2020, 9, 2301. [Google Scholar] [CrossRef]
- Furness, L.; Riley, P.; Wright, N.; Banka, S.; Eyre, S.; Jackson, A.; Briggs, T.A. Monogenic disorders as mimics of juvenile idiopathic arthritis. Pediatr. Rheumatol. Online J. 2022, 20, 44. [Google Scholar] [CrossRef]
- Yamashita, E.; Terreri, M.T.; Hilário, M.O.; Len, C.A. Prevalence of juvenile idiopathic arthritis in children aged 6 to 12 years in Embu das Artes, State of Sao Paulo, Brazil. Rev. Bras. Reumatol. 2013, 53, 542–545. [Google Scholar] [CrossRef]
- Bansal, N.; Pasricha, C.; Kumari, P.; Jangra, S.; Kaur, R.; Singh, R. A comprehensive overview of juvenile idiopathic arthritis: From pathophysiology to management. Autoimmun. Rev. 2023, 22, 103337. [Google Scholar] [CrossRef]
- Prahalad, S.; Glass, D.N. A comprehensive review of the genetics of juvenile idiopathic arthritis. Pediatr. Rheumatol. Online J. 2008, 6, 11. [Google Scholar] [CrossRef]
- Hoang, T.K.; Albert, D.A. Novel presentations of periodic fever syndromes: Discrepancies between genetic and clinical diagnoses. Eur. J. Rheumatol. 2019, 6, 12–18. [Google Scholar] [CrossRef]
- De Silvestri, A.; Capittini, C.; Poddighe, D.; Marseglia, G.L.; Mascaretti, L.; Bevilacqua, E.; Scotti, V.; Rebuffi, C.; Pasi, A.; Martinetti, M.; et al. HLA-DRB1 alleles and juvenile idiopathic arthritis: Diagnostic clues emerging from a meta-analysis. Autoimmun. Rev. 2017, 16, 1230–1236. [Google Scholar] [CrossRef]
- Li, Y.R.; Li, J.; Zhao, S.D.; Bradfield, J.P.; Mentch, F.D.; Maggadottir, S.M.; Hou, C.; Abrams, D.J.; Chang, D.; Gao, F.; et al. Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat. Med. 2015, 21, 1018–1027. [Google Scholar] [CrossRef]
- Crinzi, E.A.; Haley, E.K.; Poppenberg, K.E.; Jiang, K.; Tutino, V.M.; Jarvis, J.N. Analysis of chromatin data supports a role for CD14+ monocytes/macrophages in mediating genetic risk for juvenile idiopathic arthritis. Front. Immunol. 2022, 13, 913555. [Google Scholar] [CrossRef]
- Nigrovic, P.A.; Martínez-Bonet, M.; Thompson, S.D. Implications of juvenile idiopathic arthritis genetic risk variants for disease pathogenesis and classification. Curr. Opin. Rheumatol. 2019, 31, 401–410. [Google Scholar] [CrossRef]
- Jiang, K.; Kessler, H.; Park, Y.; Sudman, M.; Thompson, S.D.; Jarvis, J.N. Broadening our understanding of the genetics of Juvenile Idiopathic Arthritis (JIA): Interrogation of three dimensional chromatin structures and genetic regulatory elements within JIA-associated risk loci. PLoS ONE 2020, 15, e0235857. [Google Scholar] [CrossRef]
- Nazarova, L.S.h.; Danilko, K.V.; Malievsky, V.A.; Karimov, D.O.; Bakirov, A.B.; Viktorova, T.V. Interaction of immune response mediator genes in a predisposition to juvenile idiopathic arthritis. Russ. Open Med. J. 2022, 11, e0311. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.R.; Glessner, J.T.; Yang, J.; March, M.E.; Kao, C.; Vaccaro, C.N.; Bradfield, J.P.; Li, J.; Mentch, F.D.; et al. Identification of Novel Loci Shared by Juvenile Idiopathic Arthritis Subtypes Through Integrative Genetic Analysis. Arthritis Rheumatol. 2022, 74, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Bowes, J.; Yarwood, A.; Smith, S.; Tarasek, D. Genetics of Juvenile Idiopathic Arthritis: The identification of a novel risk locus and clinical subgroup analysis. Ann. Rheum. Dis. 2019, 78 (Suppl. S2), 170. [Google Scholar] [CrossRef]
- Monteiro, F.P.; Vieira, T.P.; Sgardioli, I.C.; Molck, M.C.; Damiano, A.P.; Souza, J.; Monlleó, I.L.; Fontes, M.I.; Fett-Conte, A.C.; Félix, T.M.; et al. Defining new guidelines for screening the 22q11.2 deletion based on a clinical and dysmorphologic evaluation of 194 individuals and review of the literature. Eur. J. Pediatr. 2013, 172, 927–945. [Google Scholar] [CrossRef] [PubMed]
- McDonal-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.S.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Prim. 2015, 1, 15071. [Google Scholar] [CrossRef] [PubMed]
- Smyk, M.; Geremek, M.; Ziemkiewicz, K.; Gambin, T.; Kutkowska-Kazmierczak, A.; Kowalczyk, K.; Plaskota, I.; Wiśniowiecka-Kowalnik, B.; Bartnik-Głaska, M.; Niemiec, M.; et al. Coexisting conditions modifying phenotypes of patients with 22q11.2 Deletion Syndrome. Genes 2023, 14, 680. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.M.W.; Gil-da-Silva-Lopes, V.L. An overview of the trajectory of Brazilian individuals with 22q11.2 deletion syndrome until diagnosis. Orphanet J. Rare Dis. 2022, 17, 67. [Google Scholar] [CrossRef] [PubMed]
- Zori, R.T.; Rasmussen, S.A.; Ayoub, E.M.; Gray, B.A.; Bent-Williams, A.; Stalker, H.J.; Williams, C.A. Juvenile rheumatoid arthritis (JRA) in two individuals with velo-cardio-facial syndrome (abstract). Am. J. Hum. Genet. 1995, 57, A106. [Google Scholar]
- Rasmussen, S.A.; Williams, C.A.; Ayoub, E.M.; Sleasman, J.W.; Gray, B.A.; Bent-Williams, A.; Stalker, H.J.; Zori, R.T. Juvenile rheumatoid arthritis in velo-cardio-facial syndrome: Coincidence or unusual complication? Am. J. Med. Genet. 1996, 64, 546–550. [Google Scholar] [CrossRef]
- Verloes, A.; Curry, C.; Jamar, M.; Herens, C.; O’Lague, P.; Marks, J.; Sarda, P.; Blanchet, P. Juvenile rheumatoid arthritis and del(22q11) syndrome: A non-random association. J. Med. Genet. 1998, 35, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.; Stiehm, E.R.; Woo, P.; Murray, K.J. Juvenile idiopathic polyarticular arthritis and IgA deficiency in the 22q11 deletion syndrome. J. Rheumatol. 2001, 28, 2326–2334. [Google Scholar] [PubMed]
- Pelkonen, P.; Lahdenne, P.; Lantto, R.; Honkanen, V. Chronic arthritis associated with chromosome deletion 22q11.2 syndrome. J. Rheumatol. 2002, 29, 2648–2650. [Google Scholar] [PubMed]
- Salehzadeh, F.; Bagheri, A. Association of juvenile idiopathic arthritis and digeorge syndrome; a case report. Iran. J. Pediatr. 2014, 24, 334–336. [Google Scholar] [PubMed]
- Budarf, M.; McDonald, T.; Sellinger, B.; Kozak, C.; Graham, C.; Wistow, G. Locaization of the human gene for macrophage migration inhibitory factor (MIF) to chromosome 22q11.2. Genomics 1997, 39, 235–236. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.V.C.; Mascaro-Cordeiro, B.; Lucon, D.R.; Nóbrega, M.S.; Reis, R.S.; de Alexandre, R.B.; Moura, L.M.S.; de Oliveira, G.S.; Guedes, R.L.M.; Caraciolo, M.P.; et al. The Brazilian Rare Genomes Project: Validation of Whole Genome Sequencing for Rare Diseases Diagnosis. Front. Mol. Biosci. 2022, 9, 821582. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, E.D.S.; Griffith, S.; Martin, R.; Antonescu, C.; Posey, J.E.; Coban-Akdemir, Z.; Jhangiani, S.N.; Doheny, K.F.; Lupski, J.R.; Valle, D.; et al. Variant-level matching for diagnosis and discovery: Challenges and opportunities. Hum. Mutat. 2022, 43, 782–790. [Google Scholar] [CrossRef]
- Degboe, Y.; Vastert, S.J.; Prakken, B.J.; McInnes, I.B. How does age determine the development of human immune-mediated arthritis? Nat. Rev. Rheumatol. 2022, 18, 501–512. [Google Scholar] [CrossRef]
- Cron, R.Q.; Sullivan, K.E. Chronic arthritis without uveitis in velocardiofacial syndrome. J. Pediatr. 2006, 149, 281. [Google Scholar] [CrossRef]
- Sullivan, K.E.; McDonald-McGinn, D.M.; Driscoll, D.A.; Zmijewski, C.M.; Ellabban, A.S.; Reed, L.; Emanuel, B.S.; Zackai, E.H.; Athreya, B.H.; Keenan, G. Juvenile rheumatoid arthritis-like polyarthritis in chromosome 22q11.2 deletion syndrome (DiGeorge anomalad/velocardiofacial syndrome/conotruncal anomaly face syndrome). Arthritis Rheum. 1997, 40, 430–436. [Google Scholar] [CrossRef]
- Homans, J.F.; Tromp, I.N.; Colo, D.; Schlösser, T.P.C.; Kruyt, M.C.; Deeney, V.F.X.; Crowley, T.B.; McDonald-McGinn, D.M.; Castelein, R.M. Orthopaedic manifestations within the 22q11.2 Deletion syndrome: A systematic review. Am. J. Med. Genet. A 2018, 176, 2104–2120. [Google Scholar] [CrossRef]
- Lewis, C.M.; Vassos, E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020, 12, 44. [Google Scholar] [CrossRef]
- Brown, M.A. Polygenic risk scores. Semin. Arthritis Rheum. 2024, 64S, 152330. [Google Scholar] [CrossRef]
- Wray, N.R.; Yang, J.; Goddard, M.E.; Visscher, P.M. The genetic interpretation of area under the ROC curve in genomic profiling. PLoS Genet. 2010, 6, e1000864. [Google Scholar] [CrossRef]
- Cánovas, R.; Cobb, J.; Brozynska, M.; Bowes, J.; Li, Y.R.; Smith, S.L.; Hakonarson, H.; Thomson, W.; Ellis, J.A.; Abraham, G.; et al. Genomic risk scores for juvenile idiopathic arthritis and its subtypes. Ann. Rheum. Dis. 2020, 79, 1572–1579. [Google Scholar] [CrossRef]
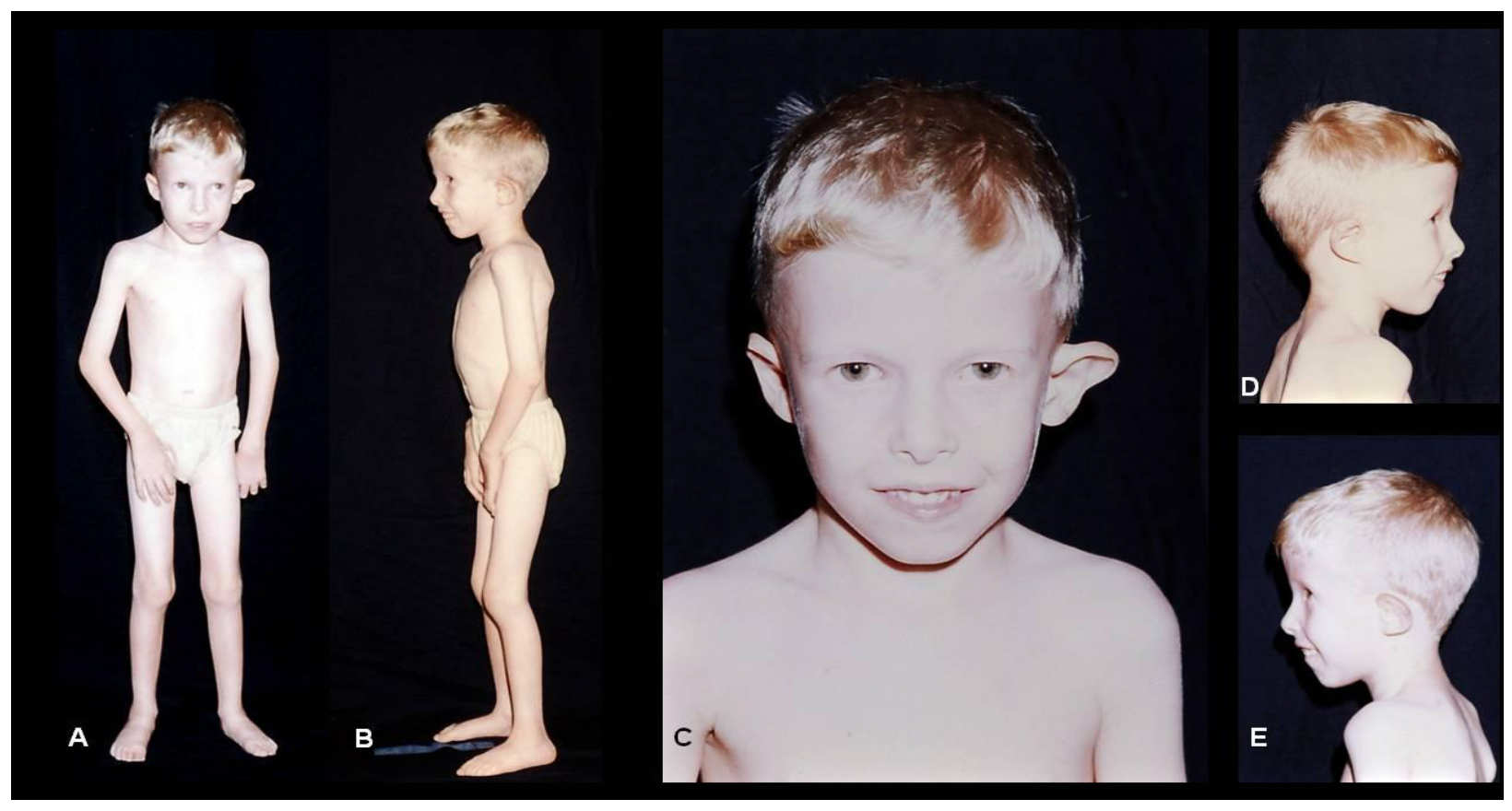



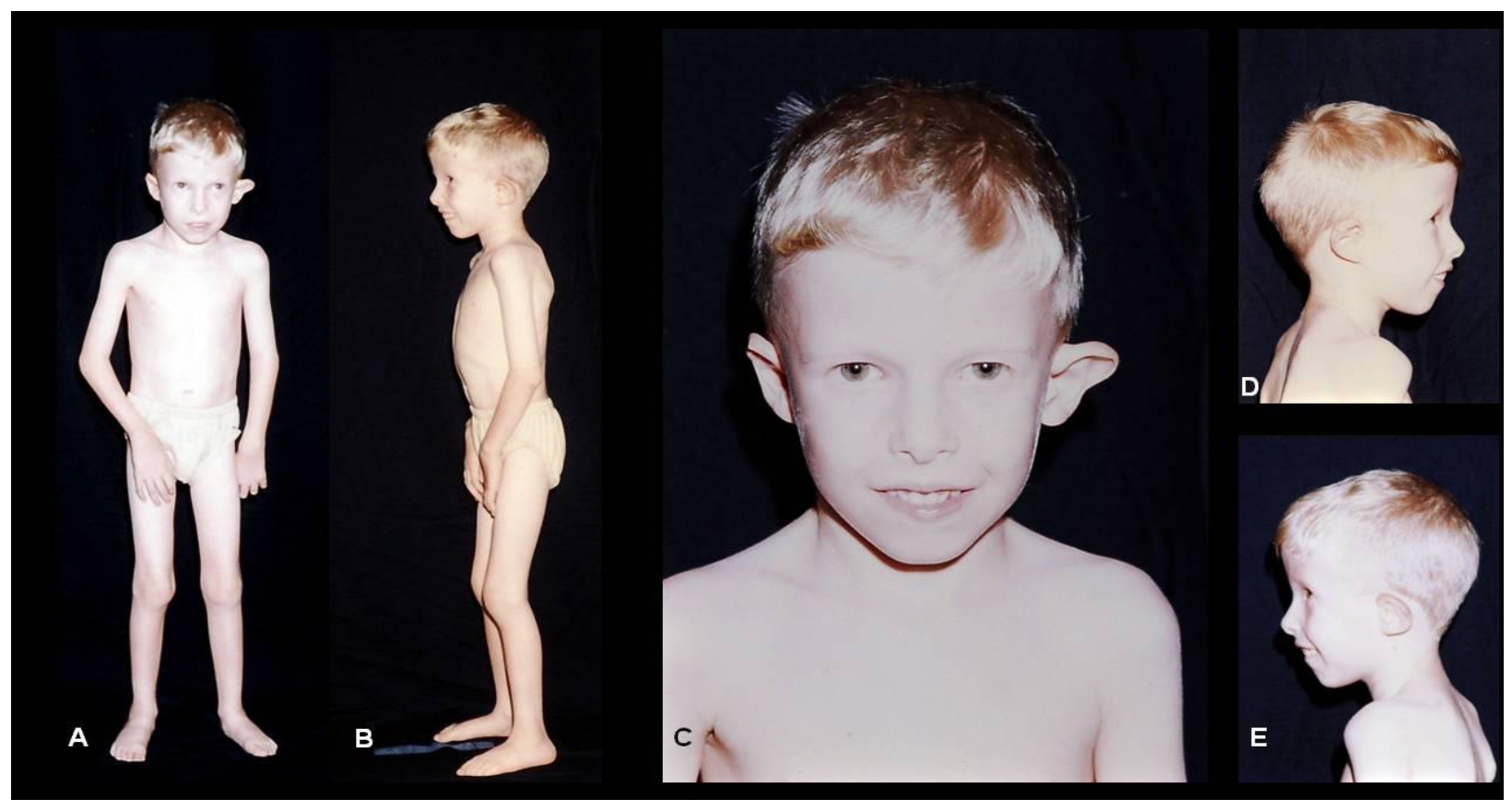{kind=link}
{kind=link}
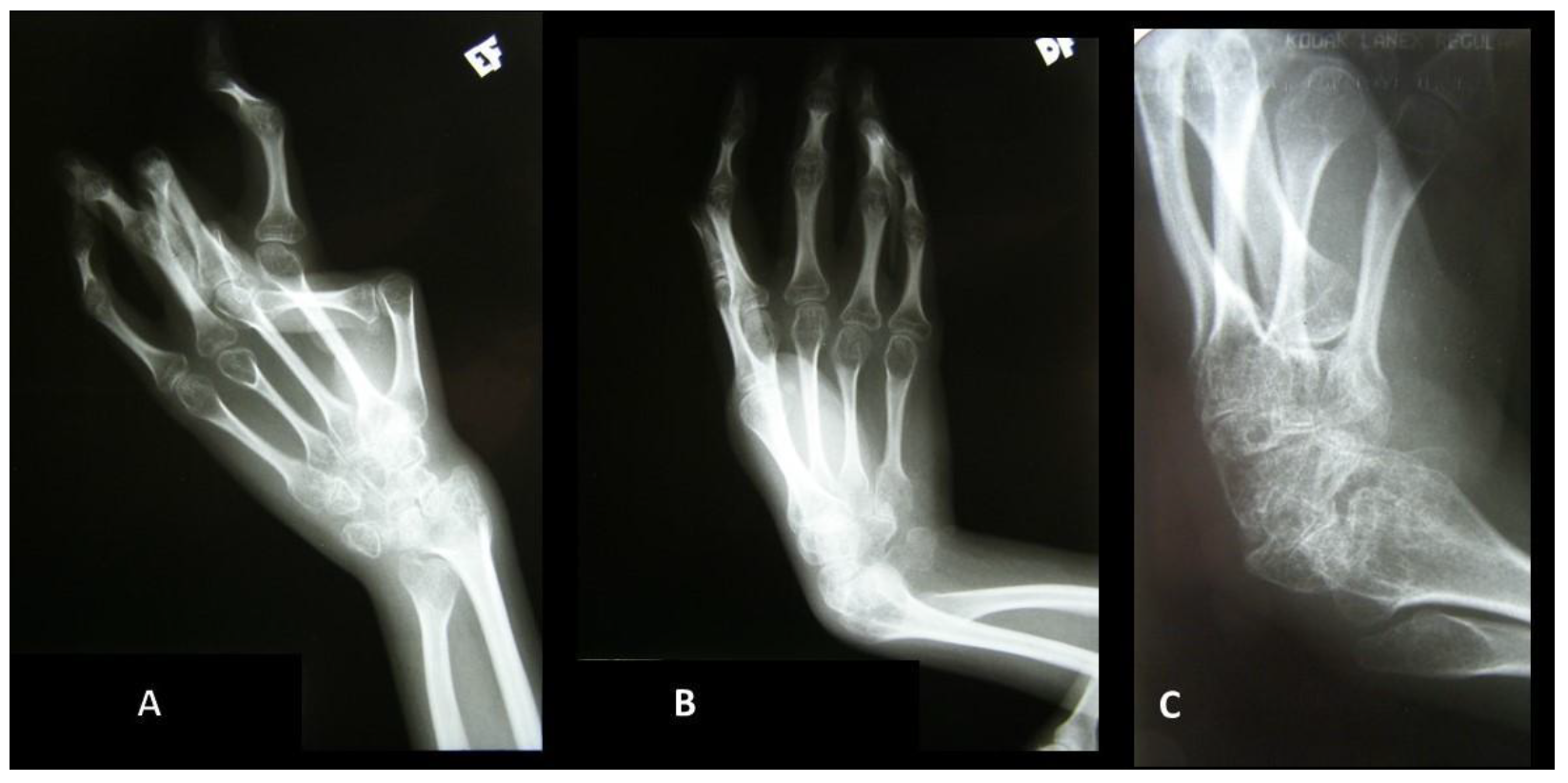
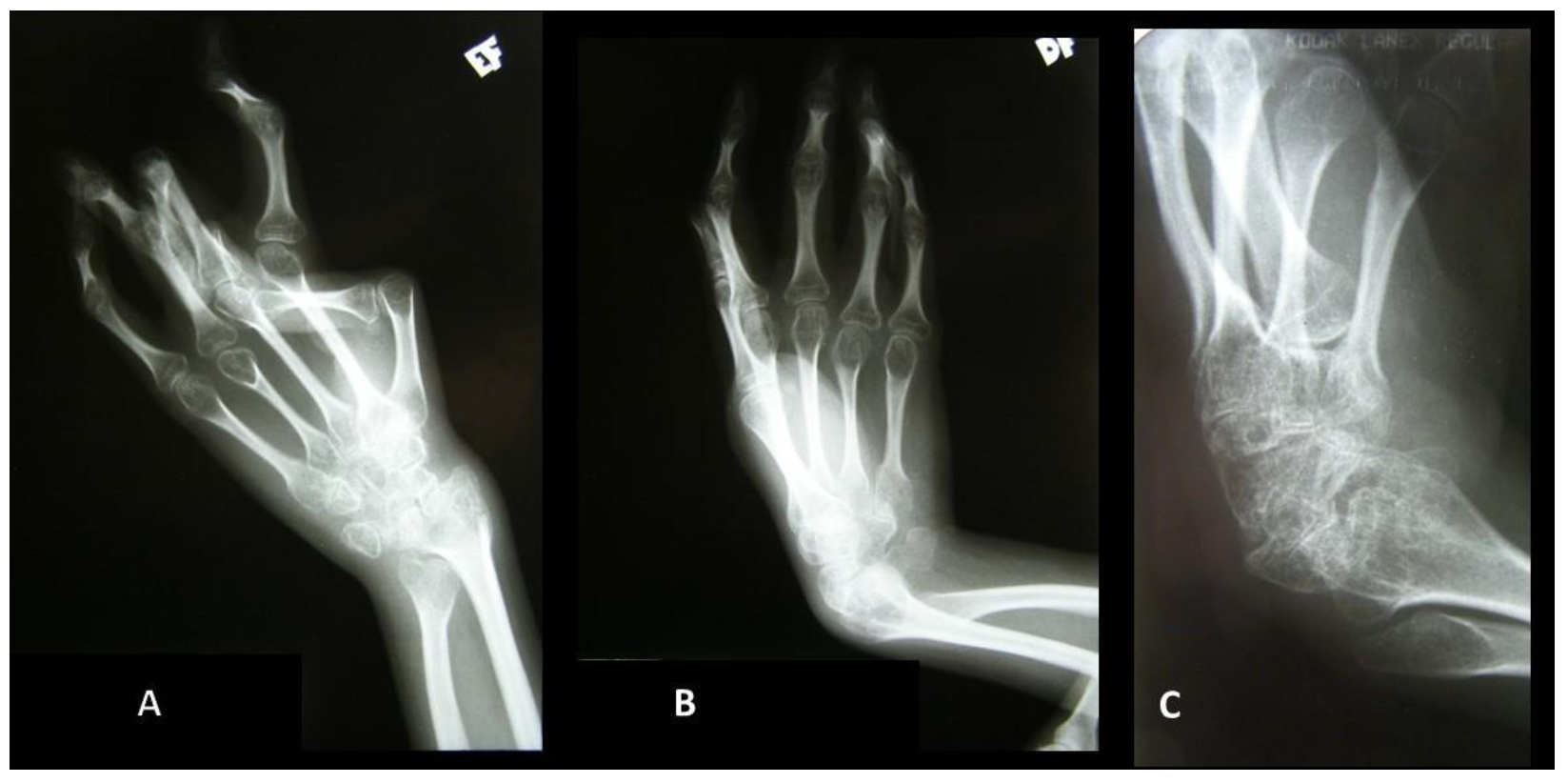{kind=link}
{kind=link}
| Gene/Position | Polymorphism | cDNA/Nucleotide | Location | Zygosity | Ref. |
|---|---|---|---|---|---|
| ADGRL2 | rs2066363 | c.-101 + 10040C > T | intron 2 | hom | [8,10] |
| AFF3<>LINC01104 | rs6740838 | n.100197037T > G | intergenic | het | [9] |
| AHI1 | rs9321502 | c.3166-11790G > T | intron 25 | het | [9] |
| ANKRD55 | rs10213692 | c.484-2493A > G | intron 7 | het | [1] |
| rs7731626 | c.484-4927C > T | intron 7 | het | [1] | |
| rs71624119 | c.484-974C > T | intron 7 | het | [9] | |
| C5<>OT1 | rs10818488 | n.1341T > C | intergenic | hom | [11] |
| rs2900180 | n.641-595A > G | intergenic | hom | [11] | |
| CLIC4<>RUNX3 | rs4648881 | n.24870664G > A | intergenic | het | [9] |
| COG6 | rs7993214 | c.1827-11560T > C | intron 19 # | hom | [9] |
| FAS | rs7069750 | c.31-410G > T | intron 2 | hom | [1] |
| HBP1 | rs111865019 | c.-16 + 2616A > G | intron 1 | het | [9] |
| IL1B | rs16944 | c.-598T > C | UTR 5′ | het | [11] |
| IL2<>IL21 | rs1479924 | n.122466445G > A | intergenic | het | [1,9] |
| IL2RA | rs706778 | c.64 + 5102G > A | intron 1 # | hom | [1] |
| IL2RB | rs2284033 | c.389-259C > T | intron 6 | het | [1,9] |
| IL6<>AS1 | rs1800795 | n.54-321G > C | intergenic | hom | [12] |
| IL6<>TOMM7 | rs7808122 | n.22758461T > C | intergenic | hom | [9] |
| rs6946509 | n.22769871T > C | intergenic | hom | [1] | |
| IL19 | rs1800872 | c.-149 + 1984T > G | intron 1 | het | [12] |
| IRF1 | rs4705862 | c.*6424T > A | UTR 3′ | het | [9] |
| LNPEP | rs27290 | c.2219 + 553G > A | intron 12 | het | [8,11] |
| rs27293 | c.2377-826A > G | intron 14 # | het | [1,11] | |
| LOC102723427<>CTN66 | rs727845 | n.68142222A > G | intergenic | hom | [11] |
| LOC105377621 | rs28362491 | n.48 + 1438_48 + 1441del | intergenic # | het | [12] |
| LTBR | rs10849448 | c.-174A > G | UTR 5′ | het | [1] |
| rs2364480 | c.516C > A (p.A172=) | exon 5 | het | [9] | |
| LURAP1L | rs7042370 | c.312 + 9047T > C | intron 1 # | hom | [13] |
| NAA25 | rs17696736 | c.1729-571T > C | intron 16 | hom | [11] |
| PADI4 | rs2240336 | c.1048-34C > T | intron 10 # | hom | [12] |
| PTH1R | rs1138518 | c.1389T > C (p.N463=) | exon 15 | het | [9] |
| PTPN2 | rs2847293 | c.*3374T > A | UTR 3′ | hom | [9] |
| RNF215 | rs5753109 | c.745-1498A > G | intron 6 | het | [9] |
| RUNX1<>LOC100506403 | rs812903 | n.35340290T > A | intergenic # | het | [1] |
| rs9979383 | n.35343463C > T | intergenic # | het | [9] | |
| SGF29 | rs497523 | c.-16 + 12513T > C | intron 1 | het | [14] |
| SNORA88<>WT1 | rs12795402 | n.32234390T > C | intergenic | hom | [13] |
| STAT4 | rs10174238 | c.274-31983C > T | intron 4 | hom | [1] |
| TENM3<>DCTD | rs7660520 | n.182824168G > A | intergenic | het | [1,10] |
| ZFP36L1 | rs3825568 | c.76-801G > A | intron 2 # | het | [1,11] |
| rs12434551 | c.*2886T > A | UTR 3′ | het | [9,11] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira-Sobrinho, R.P.; Appenzeller, S.; Holanda, I.P.; Heleno, J.L.; Jorente, J.; on behalf of the Rare Genomes Project Consortium; Vieira, T.P.; Steiner, C.E. Genome Sequencing in an Individual Presenting with 22q11.2 Deletion Syndrome and Juvenile Idiopathic Arthritis. Genes 2024, 15, 513. https://doi.org/10.3390/genes15040513
de Oliveira-Sobrinho RP, Appenzeller S, Holanda IP, Heleno JL, Jorente J, on behalf of the Rare Genomes Project Consortium, Vieira TP, Steiner CE. Genome Sequencing in an Individual Presenting with 22q11.2 Deletion Syndrome and Juvenile Idiopathic Arthritis. Genes. 2024; 15(4):513. https://doi.org/10.3390/genes15040513
Chicago/Turabian Stylede Oliveira-Sobrinho, Ruy Pires, Simone Appenzeller, Ianne Pessoa Holanda, Júlia Lôndero Heleno, Josep Jorente, on behalf of the Rare Genomes Project Consortium, Társis Paiva Vieira, and Carlos Eduardo Steiner. 2024. "Genome Sequencing in an Individual Presenting with 22q11.2 Deletion Syndrome and Juvenile Idiopathic Arthritis" Genes 15, no. 4: 513. https://doi.org/10.3390/genes15040513
APA Stylede Oliveira-Sobrinho, R. P., Appenzeller, S., Holanda, I. P., Heleno, J. L., Jorente, J., on behalf of the Rare Genomes Project Consortium, Vieira, T. P., & Steiner, C. E. (2024). Genome Sequencing in an Individual Presenting with 22q11.2 Deletion Syndrome and Juvenile Idiopathic Arthritis. Genes, 15(4), 513. https://doi.org/10.3390/genes15040513