Abstract
The mitochondrial genome (mitogenome) of Actinidia macrosperma, a traditional medicinal plant within the Actinidia genus, remains relatively understudied. This study aimed to sequence the mitogenome of A. macrosperma, determining its assembly, informational content, and developmental expression. The results revealed that the mitogenome of A. macrosperma is circular, spanning 752,501 bp with a GC content of 46.16%. It comprises 63 unique genes, including 39 protein-coding genes (PCGs), 23 tRNA genes, and three rRNA genes. Moreover, the mitogenome was found to contain 63 SSRs, predominantly mono-nucleotides, as well as 25 tandem repeats and 650 pairs of dispersed repeats, each with lengths equal to or greater than 60, mainly comprising forward repeats and palindromic repeats. Moreover, 53 homologous fragments were identified between the mitogenome and chloroplast genome (cp-genome), with the longest segment measuring 4296 bp. This study represents the initial report on the mitogenome of the A. macrosperma, providing crucial genetic materials for phylogenetic research within the Actinidia genus and promoting the exploitation of species genetic resources.
1. Introduction
Actinidia macrosperma, a naturally wild kiwi, is commonly referred to as “Cat Ginseng” due to its ability to attract cats to exploit it as a natural stimulant and as a remedy for healing wounds [1]. A. macrosperma is native to eastern and southern China, mainly found in Zhejiang, Jiangsu, Jiangxi, Guangxi, and Hubei Provinces. The plant material for this study was collected from Desheng Town, Yizhou City, Guangxi Province, on the southern coast of China (108°24′ E, 24°65′ N).
A. macrosperma is a medium-sized, deciduous climbing shrub that produces white blooms during spring (April–May) and yields orange fruits in late September. It grows wild on slopes, mountain fronts, moist forest edges, or streams below 800 m above sea level [2]. Along with A. macrosperma, there are six other kiwi fruit species rich in beneficial substances such as vitamin C, phenolic compounds, carotenoids, and antioxidant activity (AAC) [3]. Flavonoids in kiwi fruit have health-promoting properties, and certain flavonoids can inhibit the activity of the angiotensin-converting enzyme (ACE), which plays a key role in regulating arterial blood pressure [4].
In China, A. macrosperma is also considered a traditional medicinal plant [5], and the roots and stems are widely utilized in treating a spectrum of ailments, including rheumatism, abscesses, joint inflammation, leprosy, jaundice, and abnormal vaginal discharge [6]. Additionally, they have been found to be useful in the management of malignancies, particularly those affecting the digestive system, liver, and lung [7]. However, due to its high medicinal value, excessive excavation has led to a sharp decline in the wild population of this species, and it has even become extinct in some areas. Therefore, it is imperative to closely observe sustainable development and effective protection of A. macrosperma. Furthermore, the complete cp-genome of A. macrosperma has been sequenced, assembled, and characterized [1].
The mitochondrion is a semi-autonomous organelle in eukaryotic cells, characterized by a small genome known as the mitochondrial genome. It interacts with nuclear and cytoplasmic genetic material to facilitate energy conversion, serving as a biochemical apparatus. Mitochondria synthesize adenosine triphosphate (ATP) through the tricarboxylic acid cycle and oxidative phosphorylation, thereby supplying energy to cells [8]. Additionally, mitochondria play roles in information transmission, cell differentiation, and apoptosis [9]. Research indicates that mitochondria are implicated in cytoplasmic male sterility (CMS), which is maternally inherited [10]. Modern plant breeders are devoted to rearranging mitochondrial genomes to restore plant fertility [11].
The plant mitochondrial genome, despite its complexity and sequencing challenges compared to the chloroplast genome, continues to thrive [12]. By November 2023, the National Center for Biotechnology Information (NCBI) had published 397 chloroplast genomes, 2515 mitochondrial genomes, and 34 plastid genomes (source: https://www.ncbi.nlm.nih.gov/genome/browse#!/organelles/; clicked on 30 November 2023). Although plant mitogenomes typically exhibit a circular genome structure [13], their physical organization still manifested various sub-genomic structures generated by fragments and repeat sequences [14] of linear, circular, and branched structures, along with homologous recombination [15]. Recombination is vital process in DNA replication [16,17,18] for all organisms, even viruses, as it plays a role in the repair and restart of damaged replication forks. Recombination could be classified into two main types, homologous recombination (HR) and non-homologous recombination (NHR), based on its mechanism and the molecules involved [19]. HR serves as the primary route for repairing plant mitochondrial DNA [20], relying on sequences with high similarity for identification and repair. The non-homologous recombination route utilizes limited or non-sequence similarities, potentially resulting in deletions or duplications, particularly in eukaryotes [21,22]. Understanding HR and the factors associated with its regulation contribute to preventing DNA damage and maintaining the stability of the mitochondrial genome. According to the endosymbiosis theory, mitochondria have their origins in an endosymbiotic α-proteobacterium residing within a host cell derived from archaea, eventually evolving into organelles of eukaryotic cells [23]. Therefore, plant mitochondrial HR shares similarities with bacterial HR [24,25,26]. Mitochondrial HR is believed to involve recurrent and interchangeable recombination events with large repeats. The changes that did not affect mitochondrial function were retained, leading to an overall increase in mitochondrial genome size [27]. Plant mitogenomes exhibit significant evolutionary diversity in terms of size, structure, content, intracellular gene transfer (IGT), and interspecific horizontal gene transfer [28]. Nonetheless, the synonymous substitution rates of mitochondrial protein-coding genes display a comparatively greater level of conservation when juxtaposed with those observed in chloroplast and nuclear genomes [29].
Actinidiaceae, composed of three genera, Actinidia, Clematoclethra, and Saurauia [30]. In the past half-century, the Actinidiaceae family has sequenced more than four nuclear genomes (https://www.ncbi.nlm.nih.gov/genome/?term=Actinidia, clicked on 30 November 2023) and over 56 complete chloroplast genomes (https://ngdc.cncb.ac.cn/cgir/genome?input_text=actindiaceae, clicked on 30 November 2023). However, previous reports have only documented a minimal number of complete mitogenomes within this family.
In this study, the assembly and annotation of the mitogenome of A. macrosperma were completed, revealing its genomic characteristics and structural features. Repeat sequences were analyzed, and the potential for transfer of chloroplast DNA into the mitogenome was discussed. Additionally, synonymous codon usage (RSCU) was investigated, and the phylogenetic relationships were explored. The results reported in this study offer a distinctive perspective into the mitochondrial evolution of an Actinidia species. Moreover, they provide a solid foundation for the effective utilization of available genetic resources and the integration of molecular marker-assisted breeding techniques in the cultivation of A. macrosperma.
2. Materials and Methods
Plant materials, genomic DNA extraction, and sequencing. The A. macrosperma materials were originally collected from Desheng Town, Yizhou City, Guangxi Province, China (108°24′ E, 24°65′ N). Genomic DNA was extracted from fresh leaves using the CTAB method [31], followed by quantification using the Qubit fluorescence assay (Invitrogen, Carlsbad, California, USA) and NanoDrop 2000 spectrophotometer (ThermoFischerScientific, Waltham, Massachusetts, USA). DNA degradation and contamination were assessed through agarose gel electrophoresis. The DNA that passed the quality check was fragmented using an ultrasonic water bath. The fragmented DNA was then used to prepare libraries for sequencing. The DNA underwent sequencing using the Nanopore platform (PromethION, Oxford Nanopore Technologies, Oxford, UK) and Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA). The ONT long-reads generated 20.7 Gb sequencing data. Regarding the NGS short-reads, Illumina PE150 (paired-end 150 bp) sequencing was employed, resulting in 12.69 Gb of raw data, which included 84,576,750 raw reads. Both of the ONT and NGS raw reads were submitted to NCBI (SRR27379599 and SRR27379600).
Mitogenome assembly and annotation. The mitochondrial genomes were assembled from ONT reads using SMARTdenovo with its default settings [32]. In order to enhance the precision and efficacy of the mitochondrial genome sequences, the ONT and NGS clean reads underwent refinement through the utilization of minimap2/miniasm [33]. BWA (v0.1.19) [34], SAMtools (v0.1.19) [35], Racon (v1.4.20) [36] and Pilon (v1.23) [37] were utilized for aligning the ONT reads to the assembled mitogenomes. The annotation of the mitochondrial genomes was performed using Geseq (https://chlorobox.mpimp-golm.mpg.de/geseq.html; clicked on 23 October 2023) online with A. arguta’s mitogenome (GenBank:MH559343) [38] serving as the reference. Circular maps of the mitochondrial genomes were generated using Ogdraw [39]. Additionally, the assembled sequences of A. macrosperma deposited in GenBank under the accession number: OR466481 (https://www.ncbi.nlm.nih.gov/nuccore/OR466481.1/ clicked on 30 November 2023).
Repeat Sequences. The analysis of simple sequence repeats (SSRs) was conducted using MISA [40] (https://webblast.ipk-gatersleben.de/misa/, clicked on 30 November 2023) with the parameters set to ‘1-10 2-5 3-4 4-3 5-3 6-3’. Tandemly repeated sequences were identified utilizing the Repeats Finder [41] (v4.09, https://tandem.bu.edu/trf/trf.html, clicked on 30 November 2023) software with default configurations. Dispersed repeats were predicted employing REPuter [42] (https://bibiserv.cebitec.uni-bielefeld.de/reputer, clicked on 30 November 2023) with the following parameters: ‘Hamming Distance 3, Maximum Computed Repeats 5000, Minimal Repeats Size 30′, and ‘e-value cut-off of 1 × 10−5’ for filtering criterion.
Chloroplast-mitochondrion-DNA transfer. The cp-genome of A. macrosperma (MN520000.1) was acquired through the NCBI Organelle Genome Resources Database (https://www.ncbi.nlm.nih.gov/genome/browse#!/organelles/, uploaded on 16 August 2020). Detection of transferred DNA fragments between chloroplast and mitochondrion genome was carried out using BLASTN, employing specified criteria: a matching rate of ≥80%, E-value of ≤1 × 10−10, and a minimum length of ≥40 [43]. Visualization of the results was conducted utilizing the software Tbtools [44] (version: 2.041, https://github.com/CJ-Chen/TBtools-II/releases clicked on 30 November 2023), leveraging its advanced circos module.
Synteny Analyses. The software MUMmer [45] (version: 3.23, http://mummer.sourceforge.net/ clicked on 30 November 2023) was employed to align the target genome to the reference genome, establishing a broad spectrum of linear relationship between the genome. To confirm the relative positional arrangement of specific regions, BLASTN was employed with the following parameters: a matching rate of ≥85%, E-value of ≤1 × 10−5, and a minimum length of ≥100. The creation of a parallel figure was achieved through the utilization of a custom Perl script.
Phylogenetic Analyses. Phylogenetic tree construction based on the core genes of mitogenomes involved clustering protein sequences from multiple samples. The cd-hit [46,47] (version 4.6.1, https://www.bioinformatics.org/cd-hit/ clicked on 30 November 2023) was utilized for clustering, considering parameters such as identity and comparison length. The clustering of protein sequences was performed according to the software’s analysis results. Pairwise sequence alignment was conducted using criteria including an identity threshold of ≥0.4 and alignment lengths equal to or greater than * 0.4. Identification of single-copy core genes was followed by protein sequence alignment using software MUSCLE [48] (version: 3.8.31, http://www.drive5.com/muscle). The resulting data were then utilized in constructing evolutionary trees using the NJ method (Neighbor-Joining method) through TreeBeST [49] (version: 1.9.2), with 1000 bootstrap replicates. Visualization of the phylogenetic trees was achieved using the web-based tool iTOL [50] (https://itol.embl.de/ clicked on 30 November 2023).
Substitution Rate Calculation. To predict the nonsynonymous substitution rate (Ka) and synonymous substitution rate (Ks) value, the MA algorithm was employed to estimate the Ka/Ks ratios of genes in each reference species compared to A. macrosperma. Subsequently, then the Ka/Ks value for the same gene was calculated. Detection of the Ka/Ks ratios for 14 protein-coding sequences obtained from the mitogenomes were detected utilizing KaKs_calculator [51] (V2.0, https://sourceforge.net/projects/kakscalculator2/ clicked on 30 November 2023).
Codon usage bias analysis. The CodonW software (version: 1.4.4, https://codonw.sourceforge.net/ clicked on 30 November 2023) was employed to conduct codon bias analysis on the mitogenomes, generating parameters such as the effective number of codon (Nc), GC and GC3, relative synonymous codon usage (RSCU). R with ggplot2 package [52] was employed to create a box plot illustrating the Ka/Ks values and a bar plot representing the RSCU values.
3. Results
3.1. The Mitogenome Characteristics of Actinidia macrosperma
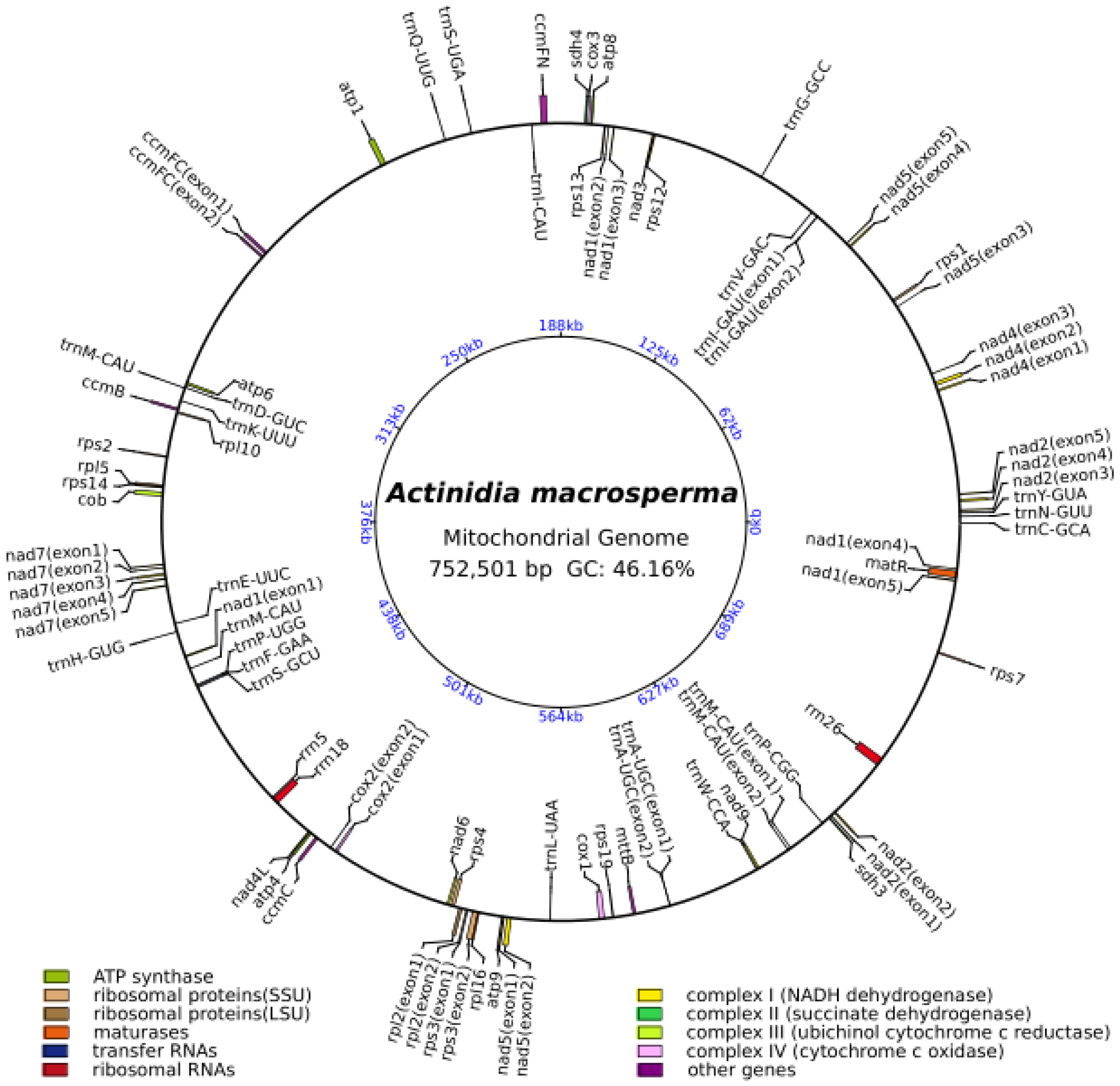
The mitogenome of A. macrosperma was reconstructed as a circular molecule comprising 752,501 base pairs (bp) (Figure 1). The annotations revealed a total of 39 protein-coding genes (PCGs), 23 tRNAs and three rRNAs in the A. macrosperma mitogenome (Tables S1 and S2). The PCGs of A. macrosperma comprise five ATP synthase genes, 10 small subunits of ribosome proteins’ genes (SSU), four large subunits of ribosome proteins’ genes (LSU), a maturase, nine NADH dehydrogenase genes (complex I), two succinate dehydrogenase genes (complex II), four cytochrome c biogenesis genes (complex III), three cytochrome c oxidase genes (complex IV), a transport membrane protein gene, and an Apocytochrome b.
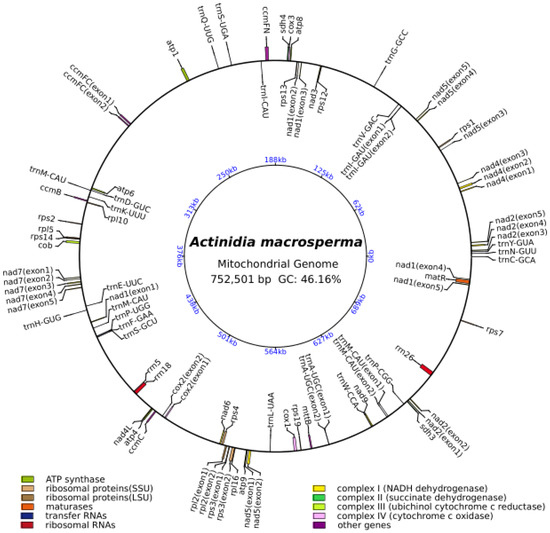
Figure 1.
The circular representation of the A. macrosperma mitogenome depicting its genomic characteristics, with the genes transcribed in a clockwise direction depicted inside the circle, while those transcribed counterclockwise were illustrated on the outside. The color scheme was assigned based on the functional classification of the genes. The innermost blue numbers represent the mitogenome scale.
3.2. Repeat Analysis
Within plant mitogenomes, a notable characteristic lies in the abundant occurrence of repetitive sequences, varying in size and dimension. These repetitions are classified into three categories: small (<50 bp), intermediate (50–500 bp), and large (>500 bp) [53]. Additionally, tandem repeats of DNA, referred to as SSRs or microsatellites, consist of 1 to 6 bp units [54]. Approximately 63 SSRs were identified within the A. macrosperma mitogenomes (Figure 2; Table S3). Predominantly, the SSRs featured a singular nucleotide repeated unit, notably A/T, constituting 55.56% of all identified SSR repeats. Nevertheless, these SSRs were evenly distributed throughout the surveyed mitogenomes. A. macrosperma harbored 35 mono-, 13 di-, 2 tri-, 11 penta-, and two hexa-repeat units. Furthermore, 25 tandem repeats were identified in the A. macrosperma mitogenomes (Figure 2; Table S4). Further investigation may explore the potential utility of these repetitive sequences in DNA fingerprinting, particularly for applications in molecular marker-assisted breeding.
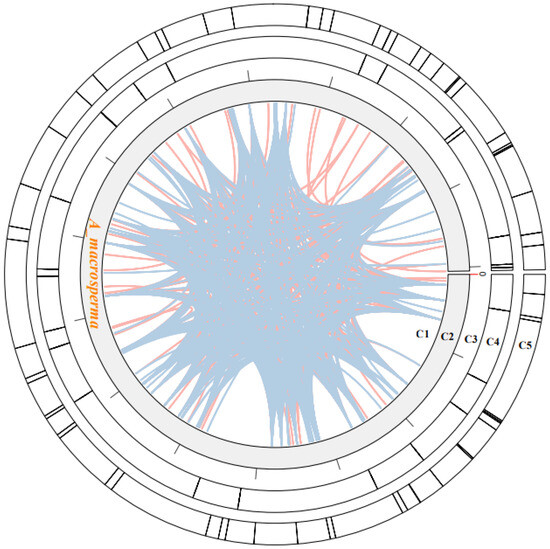
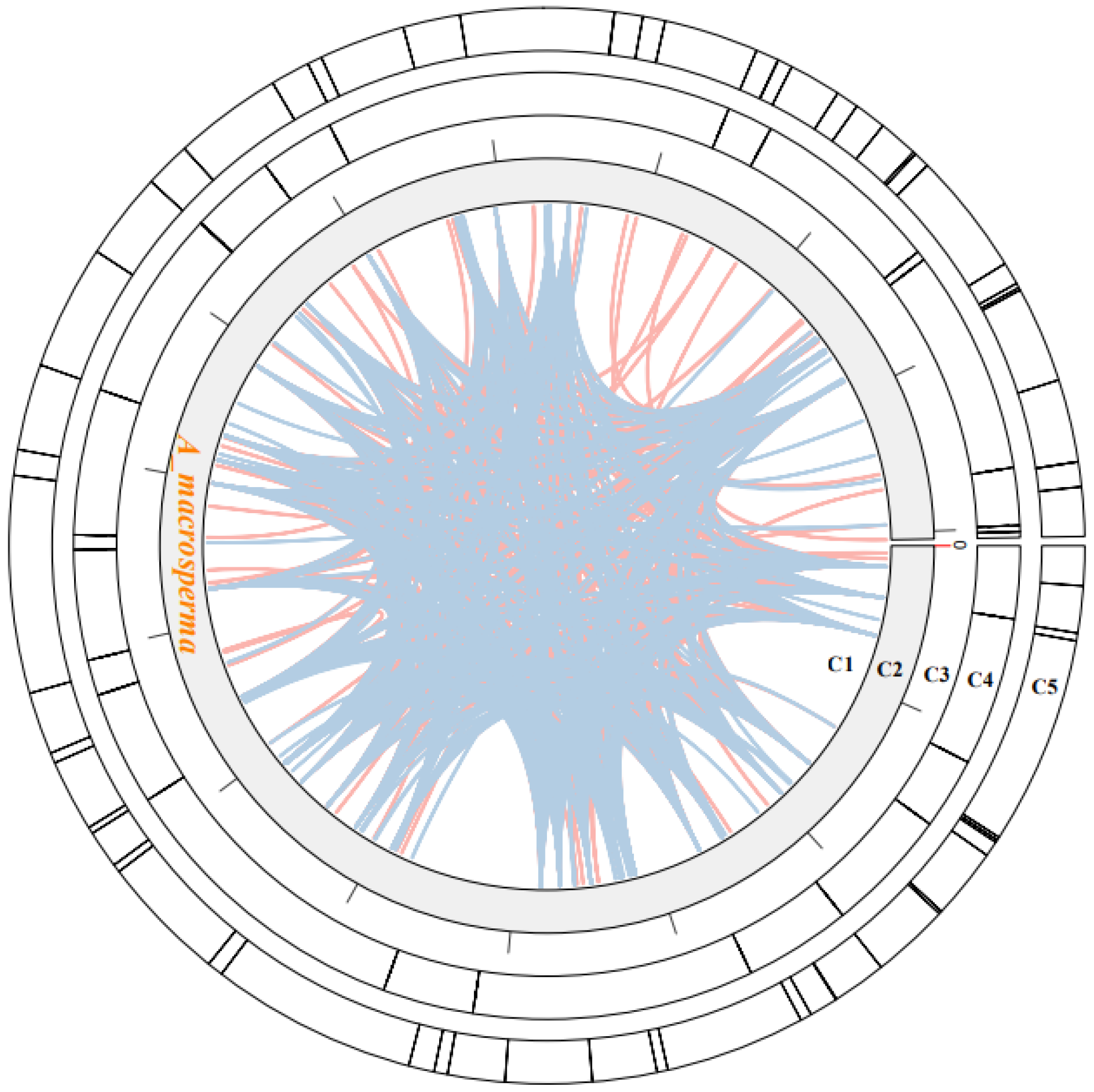
Figure 2.
The examination of repeats within the A. macrosperma mitogenomes was depicted as follows: Circle1 (C1) illustrated the dispersed repeats, where the connected blue arcs denoted forward repeats, while the pink arcs represented palindromic repeats. C2 showcased the A. macrosperma mitogenomes, with a scale of 50 kb on C3. Tandem repeats were visualized as short bars in C4. Microsatellite sequences detected by MISA were delineated in C5.
Dispersed repeats significantly contributed to the augmentation of genetic diversity, exerting a crucial influence on genome evolution [55]. Four types of dispersed repeats were identified: forward, reverse direction, complementary, and palindromic repeats [56]. Within the A. macrosperma mitogenome, forward repeats constituted 49.1% of the total repeats, while palindromic repeats accounted for 50.9%. The most extended fragments were 337 bp forward repeats from A. macrosperma (Figure 2; Table S5).
3.3. Sequence Similarity between Mitogenome and Cp-Genome
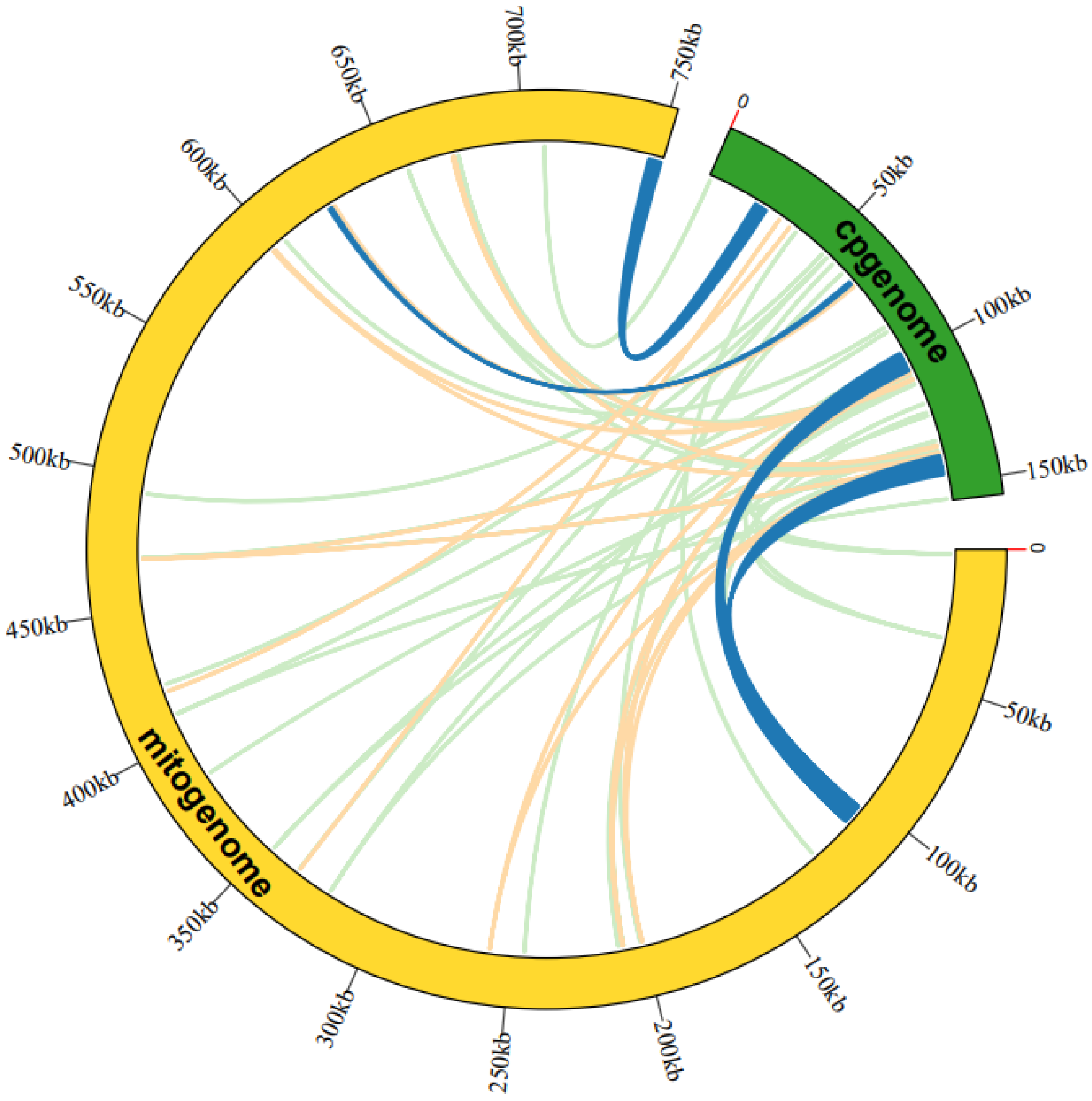
The analysis of sequence similarity indicated that the 17,783 bp sequences discovered in the A. macrosperma mitogenome likely originated from the corresponding cp-genome (Figure 3; Table S6), constituting 2.36% of the mitogenome’s sequence. A total of fifty-three homologous fragments were identified, with the longest measuring 4296 bp, between the mitogenome and cp-genome. Furthermore, these sequences’ homologous fragments encompassed eight chloroplast genes (rpoC1, ndhB, rps7, rps12, rrn16, psbF, psbE, petL) from A. macrosperma cp-genome and four mitochondrial genes (trnA-UGC, trnI-AGU, trnV-GAC, trnW-CCA) from A. macrosperma mitogenome.
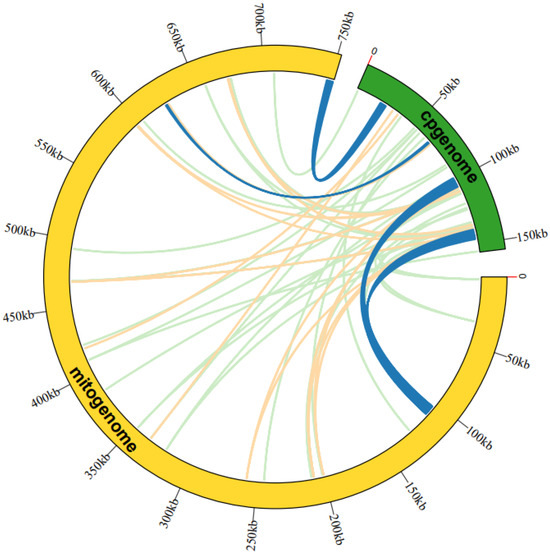
Figure 3.
An examination was undertaken regarding the transfer of mitogenome sequences from A. macrosperma cp-genomes. The outer arcs, represented in yellow and green, corresponded to the mitogenome and cp-genome, respectively, while the inner arcs depicted homologous DNA fragments. Fragments with alignment lengths exceeding 1000 bp were denoted in dark blue, those below 100 bp were depicted in jade-green, and fragments falling within this range were shown in orange. The outer arcs exhibited a scale at 50 kb intervals.
3.4. Collinearity Analyses of Mitogenome in Actinidia
The mitogenome sequences from six Actinidia species were retrieved from NCBI’s available mitogenome resources (Table S7). These genomes demonstrate comparable GC contents, ranging between 42.0% and 46.2%. However, there exists a notable diversity in genome sizes, spanning from 768,883 to 1,020,276 bp. Upon re-annotation of these mitogenomes, it was noted that certain core protein-coding genes (PCGs) were present in multiple copies, with four out of seven mitogenomes displaying re-annotated multiple-copy PCGs.
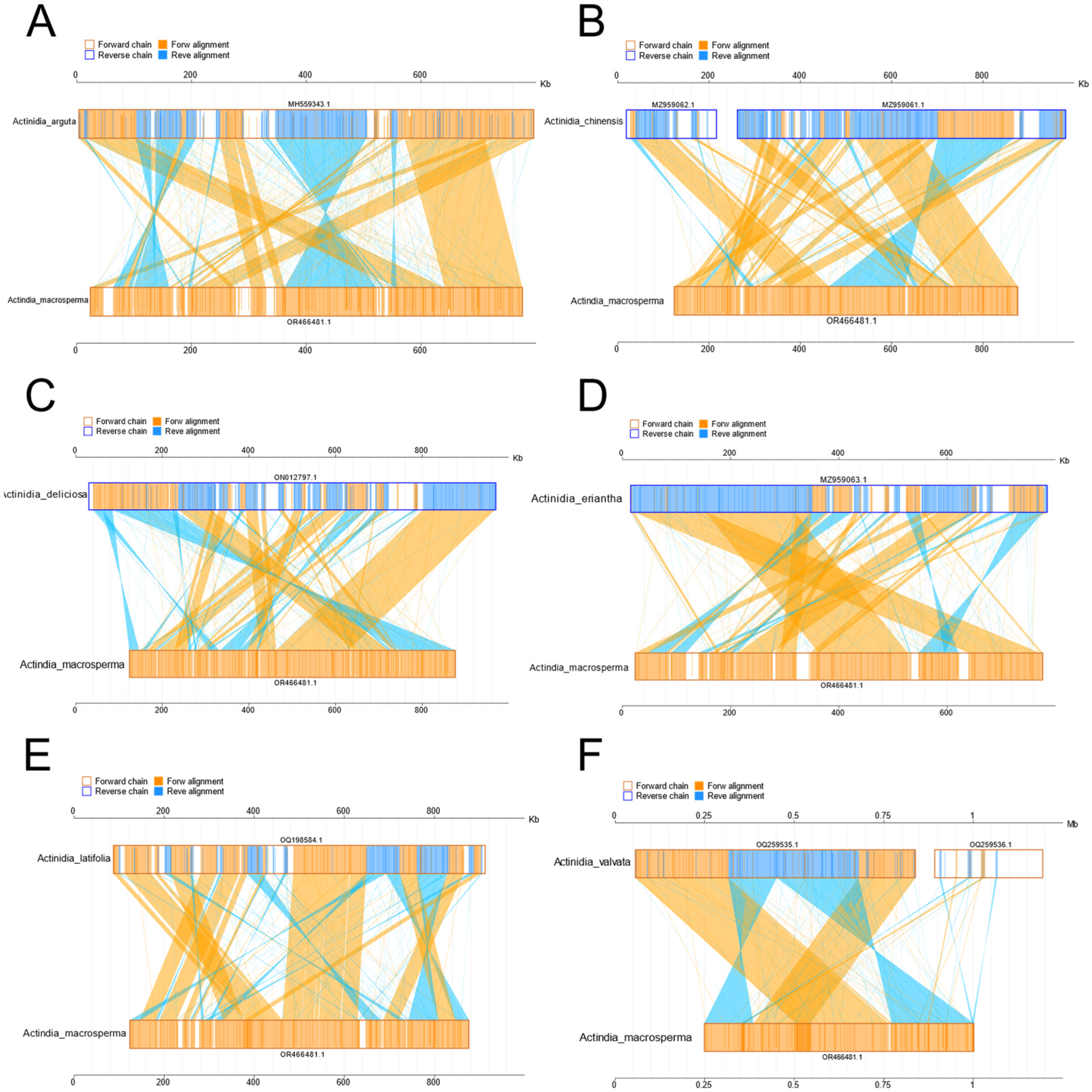
The investigation into collinearity among Actinidia mitogenomes aimed to assess genome rearrangements across different lineages. Using A. macrosperma mitogenomes as reference sequences, collinearity analyses revealed varying lengths of syntenic stretches across sampled species, ranging from less than 30 kb between A. arguta to more than 50 kb between A. valvata (Figure 4). These findings imply that significant rearrangements occur in mitochondrial genomes of Actinidia species with their divergence, leading to alterations in mitochondrial genomic synteny.
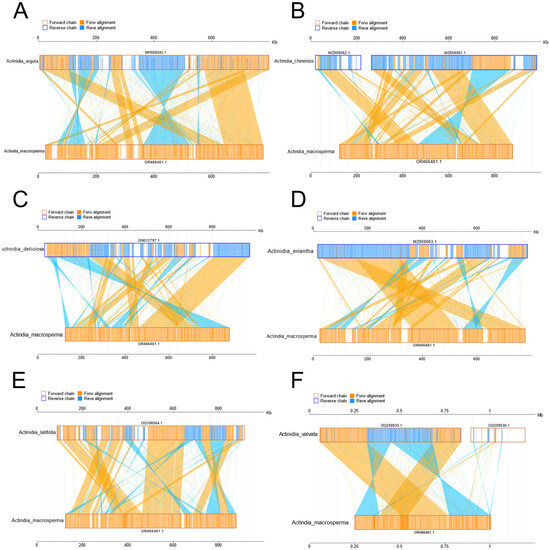
Figure 4.
The diagrams delineate collinear regions observed among disparate mitogenomes in additional Actinidia species when compared to A. macrosperma ((A): Actinidia arguta; (B): Actinidia chinensis; (C): Actinidia deliciosas; (D): Actinidia eriantha; (E): Actinidia latifolia; (F): Actinidia valvata). The outer frame colors represent the orientation of the mitogenomes sequences (orange indicating the forward direction and blue indicating the reverse direction), while the fill colors represent the alignment status (orange indicating forward alignment and blue indicating reverse alignment).
3.5. Phylogenetic Analysis
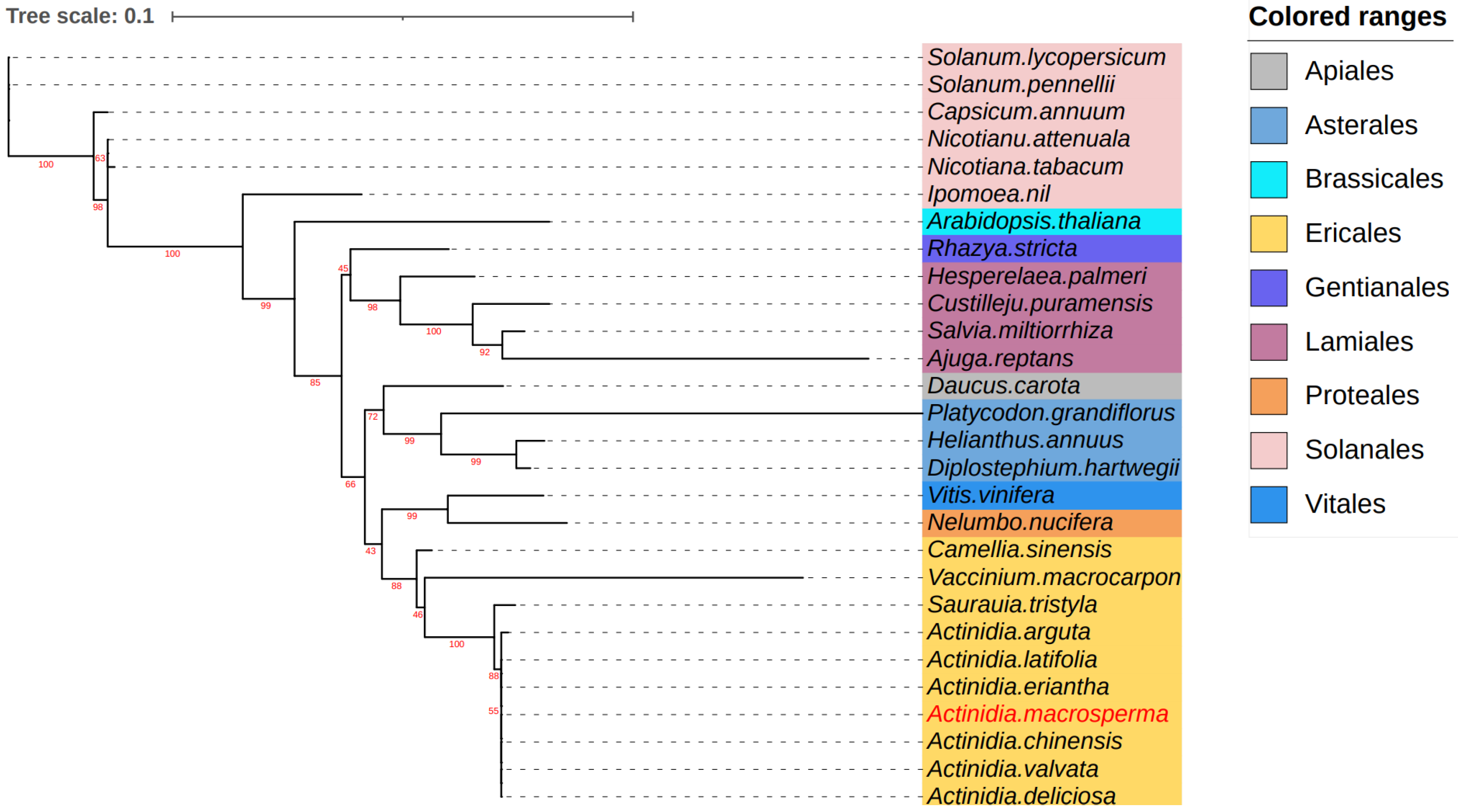
To explore the evolutionary dynamics of the A. macrosperma mitogenome, phylogenetic analyses were performed on the mitogenomes of A. latifolia, A. valvata, and 28 other related species. A set of 12 core genes (atp8, matR, ccmB, ccmFN, nad9, nad3, atp1, atp4, rps12, ccmC, nad6, cox3) was used for phylogenetic analysis. The trees generated from the core genes of mitogenomes revealed that A. macrosperma and A. chinensis, along with A. valvata and A. deliciosa, formed a cluster (Figure 5).
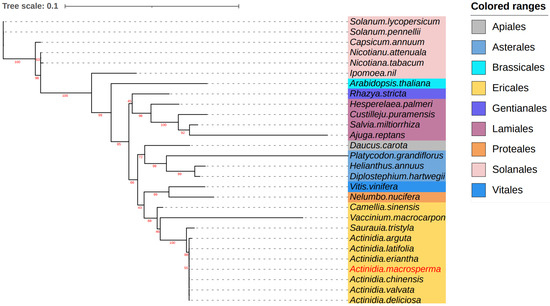
Figure 5.
The analysis delved into the phylogenetic relationships involving A. macrosperma and other species. Utilizing twelve core genes from the mitogenomes, a phylogenetic tree was constructed based on protein sequences. The mitogenomes under study were highlighted in a red font, while variously colored backgrounds were employed to denote the taxonomic classification of orders within the investigated species. The bootstrap values were denoted by the red numbers positioned on the branches of the phylogenetic tree.
3.6. Substitution Rates
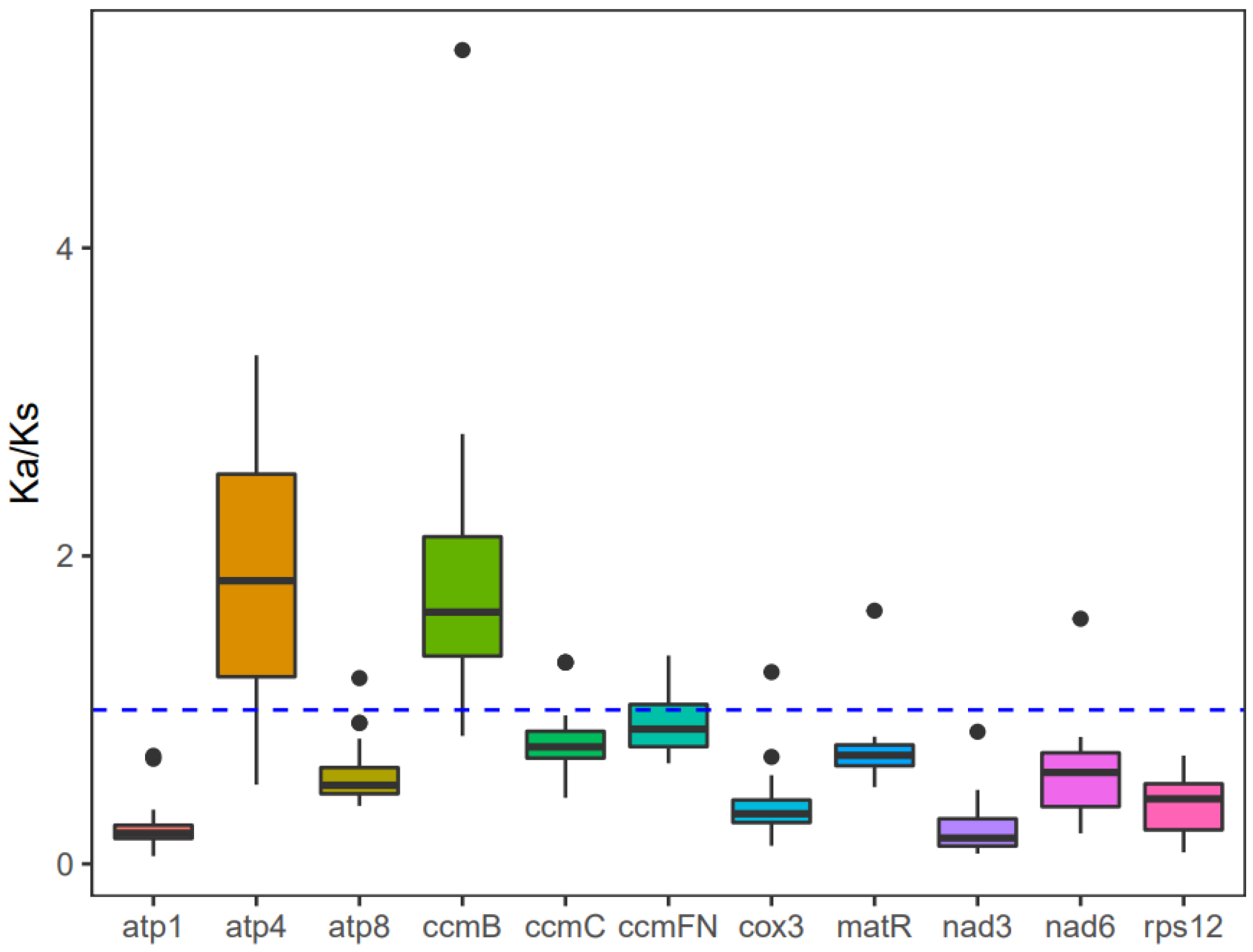
The investigation into the evolutionary rate of mitochondrial genes in A. macrosperma, Ka/Ks values were detected for 12 core protein-coding genes (Table S8). Consequently, positive selection was inferred for atp4 and ccmB due to their Ka/Ks ratios surpassing 1, while genes with lower Ka/Ks ratios were likely under purifying selection (Figure 6). Particularly, the atp1 gene displayed minimal variation and a low Ka/Ks ratio, underscoring its high conservation and pivotal role in mitogenome functionality.
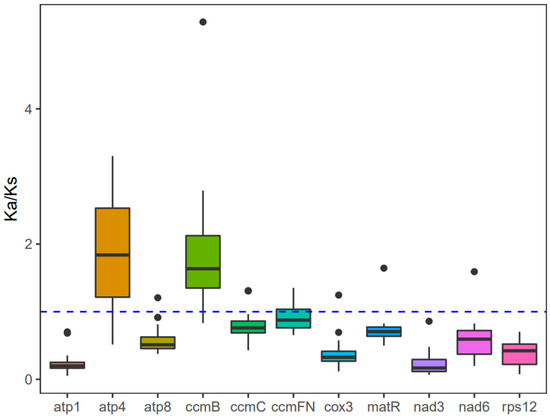
Figure 6.
The Ka/Ks values for the individual mitochondrial genes in seven species of Actinidia are illustrated in the box diagram. The x-axis and y-axis represent protein-coding genes and Ka/Ks values, respectively.
3.7. Codon Usage Bias Analysis
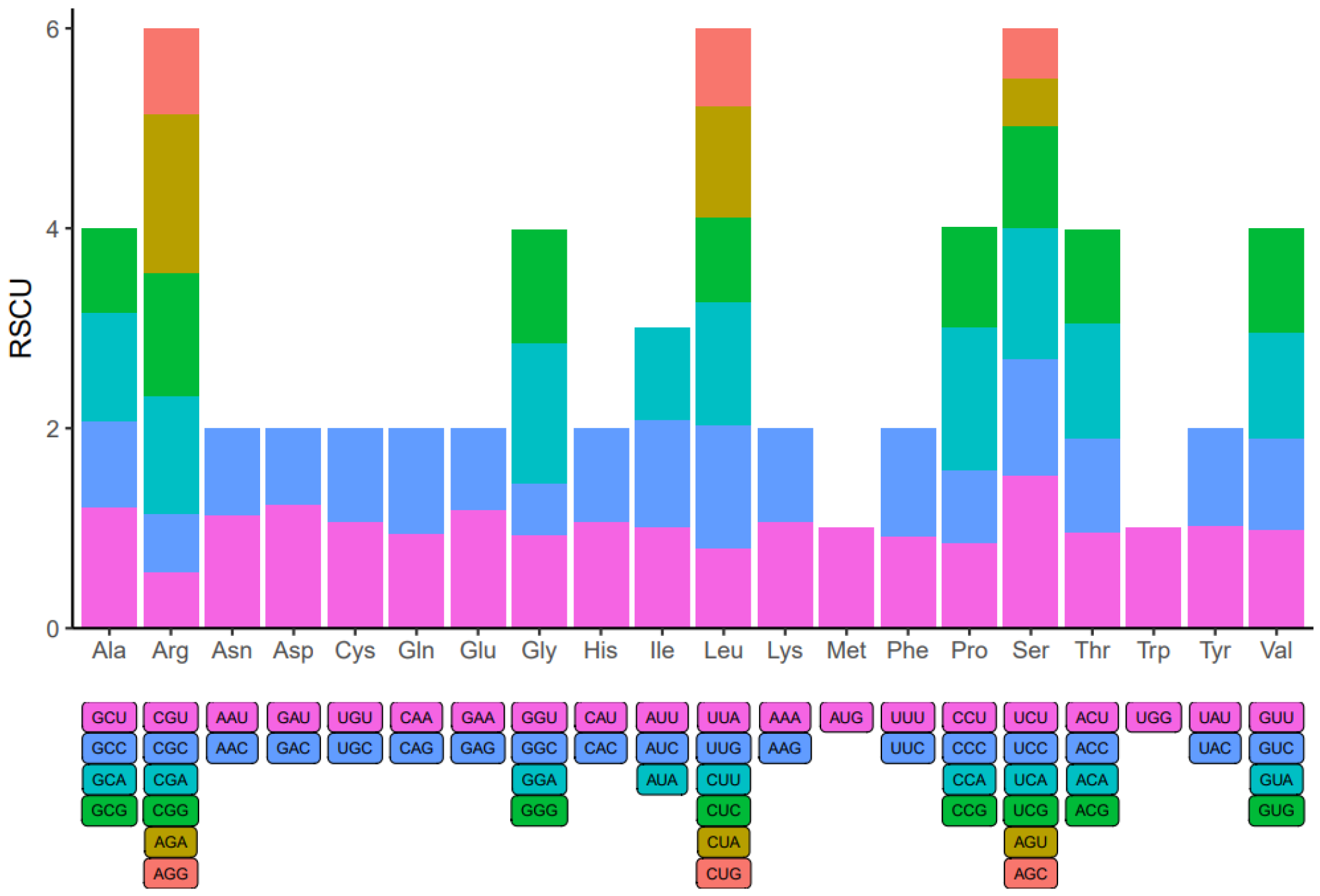
Within the genome of eukaryotic organisms, 20 different amino acids are encoded by 64 codons, with multiple codons coding for each amino acid except for Methionine and Tryptophan. Codon usage varies significantly across species due to codon degeneracy. An examination of codon usage was conducted on all mitochondrial protein-coding genes (PCGs) in A. macrosperma (Figure 7 and Table S9). Codons with a relative synonymous codon usage (RSCU) exceeding 1 were preferred by amino acids, suggesting a universal preference for codon usage among mitochondrial PCGs. For instance, Arginine (Arg) prefers AGA codons, with the maximum RSCU value among A. macrosperma mitochondrial PCGs at 1.60. Serine (Ser) closely follows, showing a preference for UCU (RSCU = 1.53). Additionally, CCA (Pro), GGA (Gly) and UCA (Ser) emerge as the three most common codons in A. macrosperma, potentially reflecting a preference shaped by long-term evolutionary selection. Moreover, in comparison to six other Actinidia species (Table S9), the result indicates that the RSCU values of A. macrosperma’s closely resemble those of A. delicese, while significant differences exist in the RSCU values of the other five species.
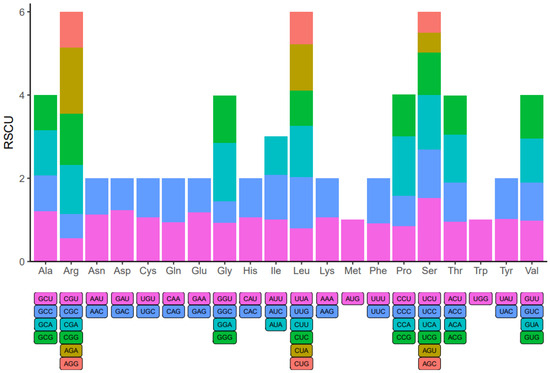
Figure 7.
Codon usage bias was observed in the mitochondrial PCGs of A. macrosperma. The term RSCU denotes relative synonymous codon usage.
4. Discussion
The present investigation achieved the successful assembly of high-quality mitogenomes for A. macrosperma by integrating sequence datasets obtained from Illumina short-reads and Oxford Nanopore long-reads. Sequence assembly elucidated that the mitogenome of A. macrosperma is represented by a single, circular molecule spanning 752,501 base pairs, displaying a GC content of 46.16% (Figure 1). Notably, the mitogenomes of A. macrosperma and the other six Actinidia species had 39 PCGs each, whether they contained one or two molecules, indicating a high conservation of protein-coding gene numbers in Actinidiaceae family mitogenomes [57,58,59]. In addition, 15 introns were found in the mitogenome of A. macrosperma, compared to the presence of 13–15 introns in the other six Actinidia species (Table S7). This evidence underscores the relative stability of mitochondrial intron content across the majority of land plant lineages, despite occasional acquisitions and numerous convergent losses over evolutionary time [60].
Repetitive elements, characterized by the presence of similar or symmetrical fragments, manifest at various loci within the genome. This phenomenon extends to both intra-species genomic regions and inter-species genomic comparisons. Studies have revealed the prevalent distribution of repeats throughout plant mitogenomes, with these sequences exhibiting limited conservation across species and a predominance of short repeats [61]. Among these, the majority of simple sequence repeats (SSRs) consist of single-nucleotide (A/T) repeats, constituting approximately 55.56% of all SSRs, likely influenced by the low GC content typical of mitochondrial genomes. This trend parallels observations in the mitogenome of A. latifolia [59]. Moreover, a significant abundance of dispersed repeats, comprising primarily forward and palindromic repeats in roughly equal proportions, was identified within the mitogenome. Similar findings have been reported in the mitochondrial genomes of Actinidia [59] and other species, such as Gleditsia sinensi [62].
Due to the unique genomic structure and evolutionary dynamics, the plant mitogenome displays heightened susceptibility to the integration of exogenous DNA [27]. Concurrently, exogenous DNA is prevalent within plant mitochondria [63]. Several investigations have highlighted notable resemblance between the mitogenome and chloroplast genome, suggesting occurrences of DNA transfer events [64,65]. These substantial homologous segments are believed to have played a pivotal role in the extensive evolutionary processes of eukaryotes, fostering genetic diversity. Additionally, studies have unveiled a bifurcated origin of tRNA genes in plant mitochondria, with a portion inherited from mitochondrial ancestors and another acquired via horizontal gene transfer (HGT) from chloroplasts [66]. The identification of chloroplast-derived tRNA genes in the A. macrosperma mitogenome has been accomplished. Remarkably, four mitogenome tRNAs, specifically trnA-UGC, trnI-GAU, trnV-GAC, and trnW-CCA, exhibited significant similarity to the entire sequence of the chloroplast genome, collectively representing 17.4 percent of all tRNAs (Table S6). Furthermore, trnW-CCA is widely distributed across angiosperm mitogenomes, exhibiting homology with chloroplasts [67].
Assessing the level of collinearity between various species can provide insights into their evolutionary divergence, with genetic relationships often inferred from collinear patterns. Collinearity analysis was undertaken to investigate mitochondrial genome DNA rearrangements across different kiwi fruit species (Figure 4). Line diagrams showed that A. macrosperma were closely related to A. valvata, with the largest collinearity region about 50 kb, while A. arguta has the smallest collinearity region, about 30 kb. The collinearity of distantly related species was relatively small, potentially resulting from significant DNA rearrangements occurring in successive generations in the past [68]. Meanwhile, evolutionary analysis based on 12 core genes also supports this conclusion in which the relationship of A. macrosperma was closer to A. valvata, A. chinensis, and A. deliciosa, while it was distantly related to A. arguta of Kiwifruit species (Figure 5). Earlier research findings was consistent with the primary conclusions mentioned above, as indicated by evolutionary studies based on the Actinidia mitogenome, showing a closer genetic distance among A. chinensis, A. deliciosa, and A. valvata, while being more distantly related to A. valvata [59].
Examining the synonymous and nonsynonymous substitution rates offers valuable understanding regarding the impact and extent of natural selection on protein evolution [69]. Through examination of the correlation between SNPs and gene mutations, comprehensive insights into genome-wide gene mutations can be obtained, enabling the inference of the evolutionary trajectory of the entire species and its underlying causes: positive selection for active adaptation to the environment, negative selection resulting from environmental pressures, or neutral selection to maintain a balance between the two. Consistent with previous research [52,59,70], the majority of mitochondrial genes underwent neutral evolution under the influence of negative selection, displaying a high degree of conservation. However, positive selection may impact genes such as atp4 and ccmB, as indicated by their dN/dS ratio surpassing 1. The protein encoded by atp4, a component of the F1F0-ATPase subunits, contributes to the conversion of proton flow into ATP within the matrix, serving as a vital energy source for cellular activities [71].The ccm gene family encoded by the ccmB gene is important for the biosynthesis of cytochrome c, which is derived from early prokaryotic cells from the plant mitotic genome [72]. Previously, it had been reported that the ccmB gene is positively selected in A. valvata [59] and Scutellaria baicalensis [73], and the gene of atp4 also exhibited highest Ka/Ks values above one in Diospyros oleifera [52]. However, the biological process for these observations is still to be explained.
5. Conclusions
The mitogenome assembly and annotation of Actinidia macrosperma, a Chinese traditional medicine plant belonging to the Actinidiaceae family, were presented for the first time. The comparative analysis encompassing gene structure, repeat regions, homologous fragments with cp-genome, and Ka/Ks codon usage played a crucial role in investigating the characteristics of the A. macrosperma mitogenome. These analyses yielded essential insights into the evolutionary history, functional characteristics, and adaptability of the A. macrosperma mitogenome, facilitating a better understanding of its distinct features. Comparative analysis, further enhanced our comprehension of the similarities and disparities between the A. macrosperma mitogenome and other related species, thereby offering deeper insights into its evolutionary role and mechanisms of ecological adaptation. In this study, the mitochondrial genome of A. macrosperma was thoroughly explored from the perspectives of genomic functional structure and genetic evolution, thus contributing valuable genetic resources for phylogenetic investigations and laying the foundation for understanding the evolutionary relationships within the Actinidiaceae family.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/genes15040514/s1, Table S1. Gene composition in the mitogenome of Actinidia macrosperma. Table S2. Gene information in the mitogenome of Actinidia macrosperma. Table S3. Microsatellite repeats in the Actinidia macrosperma mitogenome. Table S4. Tandem repeats in the Actinidia macrosperma mitogenome. Table S5. Dispersed repeats in the Actinidia macrosperma mitogenome. Table S6. Length of transferred DNA from Actinidia macrosperma cp-genome. Table S7. Features of seven mitogenomes belonging to the Actinidia. Table S8. The Ka and Ks for 12 core PCGs with Reference to Actinidia macrosperma. Table S9. The RSCU values with 39 PCGs of the Actinidia.
Author Contributions
J.G. designed the experiment and carried out the bioinformatics analyses, J.Y. collected the plant materials, J.G. and J.Y. wrote the manuscript, Y.L. collected dataset, W.S. and T.P. corrected manuscripts and administrated project. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The accession number generated for this study is available on NCBI. The raw sequencing data have been deposited in NCBI (https://www.ncbi.nlm.nih.gov/, Clicked on 23 December 2023) with accession numbers: PRJNA1056159, SAMN39090875, SRR27379599 and SRR27379600 (https://www.ncbi.nlm.nih.gov/sra?LinkName=biosample_sra&from_uid=39090875). And the assembled sequences accession number of A. macrosperma mitogenome is OR466481 (https://www.ncbi.nlm.nih.gov/nuccore/OR466481.1/).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chen, Y.; Xu, Y.; Zhang, K.; Song, Y.; He, Q.; Qian, Q.; Xu, J. The complete chloroplast genome of Actinidia macrosperma. Mitochondrial DNA Part B 2019, 4, 4188–4189. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.Y.; Wang, W.X.; Wang, W.Z.; Lu, L.Y.; Fu, F.C.; Chen, C.S. Essential oil of Actinidia macrosperma, a catnip response kiwi endemic to China. J. Zhejiang Univ. Sci. B 2006, 7, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Latocha, P.; Krupa, T.; Wołosiak, R.; Worobiej, E.; Wilczak, J. Antioxidant activity and chemical difference in fruit of different Actinidia sp. Int. J. Food Sci. Nutr. 2010, 61, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Hettihewa, S.K.; Hemar, Y.; Rupasinghe, H.V. Flavonoid-rich extract of Actinidia macrosperma (a wild kiwifruit) inhibits angiotensin-converting enzyme in vitro. Foods 2018, 7, 146. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhao, Y.P.; Wang, Z.C.; Chen, S.Y.; Fu, C.X. Composition and antimicrobial activity of the essential oil of Actinidia macrosperma from China. Nat. Prod. Res. 2007, 21, 227–233. [Google Scholar] [PubMed]
- Lai, P.F.; Zhang, H.Y. The research progress of TCM Cat Ginseng in Zhejiang province location. J. Zhejiang Coll. TCM 2002, 26, 77–78. [Google Scholar]
- Yao, G.; Wang, T.S. Medicinal plants of Actinidia genus in East China. Chin. Med. Mater. 1989, 12, 15–16. [Google Scholar]
- Daloso, D.M.; Müller, K.; Obata, T.; Florian, A.; Tohge, T.; Bottcher, A.; Riondet, C.; Bariat, L.; Carrari, F.; Nunes-Nesi, A.; et al. Thioredoxin, a master regulator of the tricarboxylic acid cycle in plant mitochondria. Proc. Natl. Acad. Sci. USA 2015, 112, E1392–E1400. [Google Scholar] [CrossRef]
- Teresa, V. Mitochondrial biogenesis: Pharmacological approaches. Curr. Pharm. Des. 2014, 20, 5507–5509. [Google Scholar]
- Levings, C., III; Pring, D.R. Restriction endonuclease analysis of mitochondrial DNA from normal and Texas cytoplasmic male-sterile maize. Science 1976, 193, 158–160. [Google Scholar] [CrossRef]
- Guo, J.; Wang, P.; Cheng, Q.; Sun, L.; Wang, H.; Wang, Y.; Kao, L.; Li, Y.; Qiu, T.; Yang, W. Proteomic analysis reveals strong mitochondrial involvement in cytoplasmic male sterility of pepper (Capsicum annuum L.). J. Proteom. 2017, 168, 15–27. [Google Scholar] [CrossRef]
- Bi, C.; Lu, N.; Xu, Y.; He, C.; Lu, Z. Characterization and analysis of the mitochondrial genome of common bean (Phaseolus vulgaris) by comparative genomic approaches. Int. J. Mol. Sci. 2020, 21, 3778. [Google Scholar] [CrossRef]
- Cheng, Y.; He, X.; Priyadarshani, S.; Wang, Y.; Ye, L.; Shi, C.; Ye, K.; Zhou, Q.; Luo, Z.; Deng, F. Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genom. 2021, 22, 167. [Google Scholar] [CrossRef]
- Liu, D.; Guo, H.; Zhu, J.; Qu, K.; Chen, Y.; Guo, Y.; Ding, P.; Yang, H.; Xu, T.; Jing, Q. Complex physical structure of complete mitochondrial genome of Quercus acutissima (Fagaceae): A significant energy plant. Genes 2022, 13, 1321. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef]
- Kreuzer, K.N. Recombination-dependent DNA replication in phage T4. Trends Biochem. Sci. 2000, 25, 165–173. [Google Scholar] [CrossRef]
- Rocha, E.P.C.; Cornet, E.; Michel, B. Comparative and evolutionary analysis of the bacterial homologous recombination systems. PLoS Genet. 2005, 1, e15. [Google Scholar] [CrossRef]
- Heyer, W.-D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef]
- Yeeles, J.T.; Poli, J.; Marians, K.J.; Pasero, P. Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 2013, 5, a012815. [Google Scholar] [CrossRef]
- Cappadocia, L.; Marechal, A.; Parent, J.-S.; Lepage, E.; Sygusch, J.; Brisson, N. Crystal structures of DNA-Whirly complexes and their role in Arabidopsis organelle genome repair. Plant Cell. 2010, 22, 1849–1867. [Google Scholar] [CrossRef]
- Puchta, H. Double-strand break-induced recombination between ectopic homologous sequences in somatic plant cells. Genetics 1999, 152, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The origin and diversification of mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, A.C.; Song, D.; Alvarez, L.A.; Wall, M.K.; Almond, D.; McClellan, D.A.; Maxwell, A.; Nielsen, B.L. Characterization of a mitochondrially targeted single-stranded DNA-binding protein in Arabidopsis thaliana. Mol. Genet. Genom. 2005, 273, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Morimatsu, K.; Kowalczykowski, S.C. RecQ helicase and RecJ nuclease provide complementary functions to resect DNA for homologous recombination. Proc. Natl. Acad. Sci. USA 2014, 111, E5133–E5142. [Google Scholar] [CrossRef] [PubMed]
- García-Medel, P.L.; Baruch-Torres, N.; Peralta-Castro, A.; Trasviña-Arenas, C.H.; Torres-Larios, A.; Brieba, L.G. Plant organellar DNA polymerases repair double-stranded breaks by microhomology-mediated end-joining. Nucleic Acids Res. 2019, 47, 3028–3044. [Google Scholar] [CrossRef] [PubMed]
- Christensen, A.C. Plant mitochondrial genome evolution can be explained by DNA repair mechanisms. Genome Biol. Evol. 2013, 5, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Gualberto, J.M.; Newton, K.J. Plant mitochondrial genomes: Dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 2017, 68, 225–252. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, K.H.; Li, W.-H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef]
- He, Z.; Zhang, X.; Zhong, Y.; Ye, L. Phylogenetic relationships of Actinidia and related genera based on micromorphological characters of foliar trichomes. Genet. Resour. Crop Evol. 2000, 47, 627–639. [Google Scholar] [CrossRef]
- Arseneau, J.R.; Steeves, R.; Laflamme, M. Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Mol. Ecol. Resour. 2017, 17, 686–693. [Google Scholar] [CrossRef]
- Liu, H.; Wu, S.; Li, A.; Ruan, J. SMARTdenovo: A de novo assembler using long noisy reads. GigaByte 2021, 2021, gigabyte15. [Google Scholar] [CrossRef]
- Li, H. Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 2016, 32, 2103–2110. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Nandi, T.; Ong, C.; Singh, A.P.; Boddey, J.; Atkins, T.; Sarkar-Tyson, M.; Essex-Lopresti, A.E.; Chua, H.H.; Pearson, T.; Kreisberg, J.F. A genomic survey of positive selection in Burkholderia pseudomallei provides insights into the evolution of accidental virulence. PLoS Pathog. 2010, 6, e1000845. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating γ-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Xu, Y.; Dong, Y.; Cheng, W.; Wu, K.; Gao, H.; Liu, L.; Xu, L.; Gong, B. Characterization and phylogenetic analysis of the complete mitochondrial genome sequence of Diospyros oleifera, the first representative from the family Ebenaceae. Heliyon 2022, 8, e09870. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Smyth, D.R. Dispersed repeats in plant genomes. Chromosoma 1991, 100, 355–359. [Google Scholar] [CrossRef]
- Ni, Y.; Li, J.; Chen, H.; Yue, J.; Chen, P.; Liu, C. Comparative analysis of the chloroplast and mitochondrial genomes of Saposhnikovia divaricata revealed the possible transfer of plastome repeat regions into the mitogenome. BMC Genom. 2022, 23, 570. [Google Scholar] [CrossRef]
- Wang, S.; Li, D.; Yao, X.; Song, Q.; Wang, Z.; Zhang, Q.; Zhong, C.; Liu, Y.; Huang, H.J. Evolution and diversification of kiwifruit mitogenomes through extensive whole-genome rearrangement and mosaic loss of intergenic sequences in a highly variable region. Genome Biol Evol. 2019, 11, 1192–1206. [Google Scholar] [CrossRef]
- Yang, J.; Ling, C.; Zhang, H.; Hussain, Q.; Lyu, S.; Zheng, G.; Liu, Y. A Comparative Genomics Approach for Analysis of Complete Mitogenomes of Five Actinidiaceae Plants. Genes 2022, 13, 1827. [Google Scholar] [CrossRef]
- Ren, W.; Wang, L.; Feng, G.; Tao, C.; Liu, Y.; Yang, J. High-quality assembly and comparative analysis of Actinidia latifolia and A. valvata mitogenomes. Genes 2023, 14, 863. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P. Variation in protein gene and intron content among land plant mitogenomes. Mitochondrion 2020, 53, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Wynn, E.L.; Christensen, A.C. Repeats of unusual size in plant mitochondrial genomes: Identification, incidence and evolution. G3 (Bethesda) 2019, 9, 549–559. [Google Scholar] [CrossRef]
- Yang, H.; Li, W.; Yu, X.; Zhang, X.; Zhang, Z.; Liu, Y.; Wang, W.; Tian, X. Insights into molecular structure, genome evolution and phylogenetic implication through mitochondrial genome sequence of Gleditsia sinensis. Sci. Rep. 2021, 11, 14850. [Google Scholar] [CrossRef]
- Alverson, A.J.; Wei, X.; Rice, D.W.; Stern, D.B.; Barry, K.; Palmer, J.D. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 2010, 27, 1436–1448. [Google Scholar] [CrossRef]
- Smith, D.R. Extending the limited transfer window hypothesis to inter-organelle DNA migration. Genome Biol. Evol. 2011, 3, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Gao, C.; Liu, J. Complete mitochondrial genomes of three Mangifera species, their genomic structure and gene transfer from chloroplast genomes. BMC Genom. 2022, 23, 147. [Google Scholar] [CrossRef]
- Sprinzl, M.; Horn, C.; Brown, M.; Ioudovitch, A.; Steinberg, S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998, 26, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.W.; Alverson, A.J.; Richardson, A.O.; Young, G.J.; Sanchez-Puerta, M.V.; Munzinger, J.; Barry, K.; Boore, J.L.; Zhang, Y.; DePamphilis, C.W.; et al. Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 2013, 342, 1468–1473. [Google Scholar] [CrossRef]
- Abrouk, M.; Murat, F.; Pont, C.; Messing, J.; Jackson, S.; Faraut, T.; Tannier, E.; Plomion, C.; Cooke, R.; Feuillet, C. Palaeogenomics of plants: Synteny-based modelling of extinct ancestors. Trends Plant Sci. 2010, 15, 479–487. [Google Scholar] [CrossRef]
- Kimura, M. Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature 1977, 267, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, S.; Chen, J.; Chen, C.; Lin, X.; Peng, H.; Zhao, Q.; Wang, X. Characterization and phylogenetic analysis of the complete mitochondrial genome sequence of Photinia serratifolia. Sci. Rep. 2023, 13, 770. [Google Scholar] [CrossRef] [PubMed]
- Clifton, S.W.; Minx, P.; Fauron, C.M.-R.; Gibson, M.; Allen, J.O.; Sun, H.; Thompson, M.; Barbazuk, W.B.; Kanuganti, S.; Tayloe, C.; et al. Sequence and comparative analysis of the maize NB mitochondrial genome. Plant Physiol. 2004, 136, 3486–3503. [Google Scholar] [CrossRef]
- Giegé, P.; Grienenberger, J.; Bonnard, G. Cytochrome c biogenesis in mitochondria. Mitochondrion 2008, 8, 61–73. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Shan, Y.; Pei, X.; Yong, S.; Liu, C.; Yu, J. Assembly of the complete mitochondrial genome of an endemic plant, Scutellaria tsinyunensis, revealed the existence of two conformations generated by a repeat-mediated recombination. Planta 2021, 254, 36. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).