
Cardiac Remodeling and Ventricular Pacing: From Genes to Mechanics
 , ,
, , 
Abstract
:1. Introduction
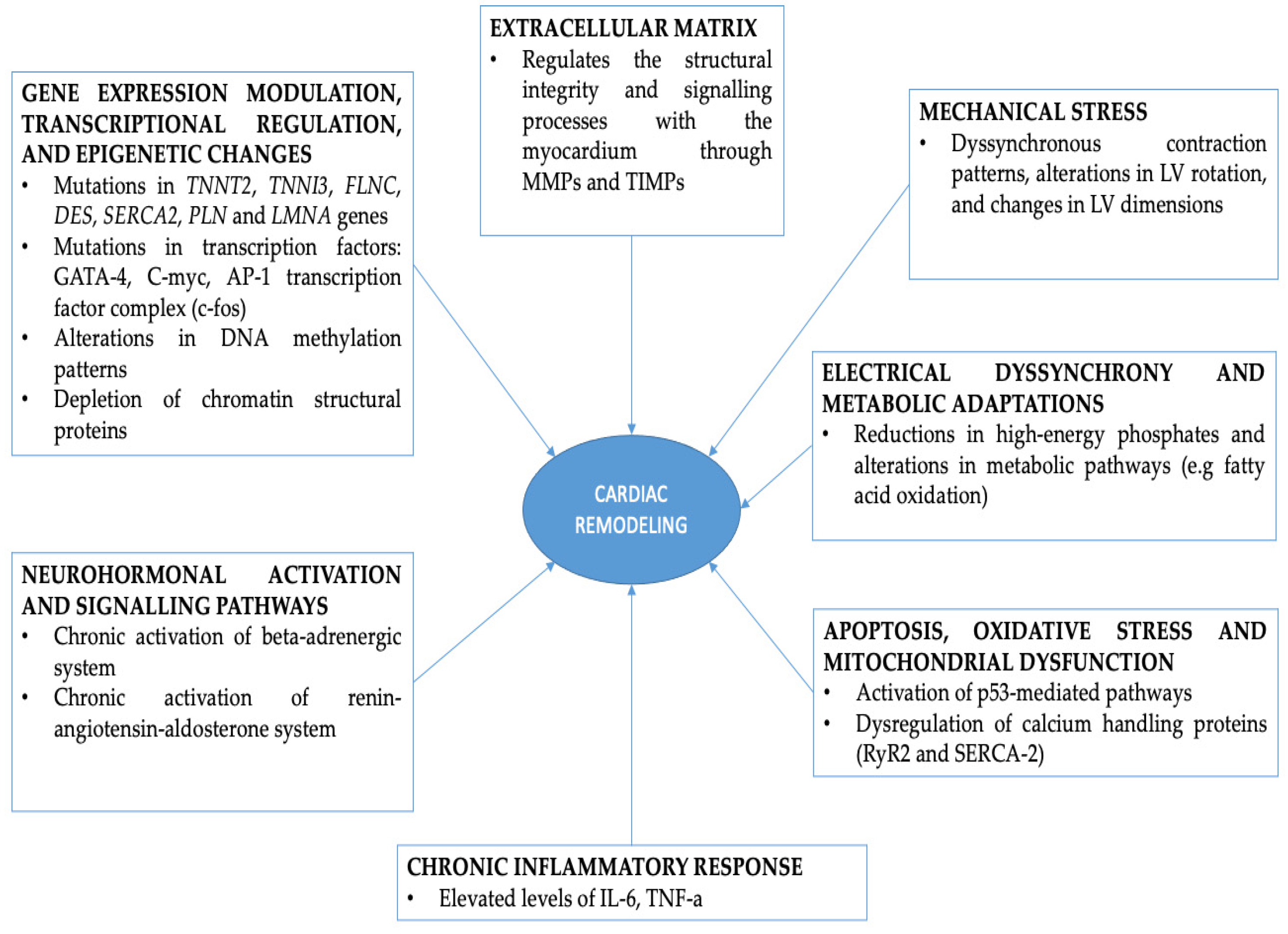
2. Transcriptional Regulation and Gene Expression Modulation during RVAP
3. Molecular Pathways Involved in Cardiac Remodeling during RVAP
3.1. Neurohormonal Activation and Dignaling Pathways
3.2. Apoptosis, Oxidative Stress, Calcium Handling Abnormalities and Mitochondrial Dysfunction
3.3. Extracellular Matrix Remodeling and Inflammatory Response during RVAP
3.4. Electical Dyssynchrony, Metabolic Adaptations and Energetics
4. Mechanical Stress and Structural Remodeling
5. Conclusions
6. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Defaye, P.; Biffi, M.; El-Chami, M.; Boveda, S.; Glikson, M.; Piccini, J.; Vitolo, M. Cardiac pacing and lead devices management: 25 years of research at EP Europace journal. Europace 2023, 25, euad202. [Google Scholar] [CrossRef] [PubMed]
- Aquilina, O. A brief history of cardiac pacing. Images Paediatr. Cardiol. 2006, 8, 17–81. [Google Scholar] [PubMed]
- Proclemer, A.; Zecchin, M.; D’Onofrio, A.; Boriani, G.; Facchin, D.; Rebellato, L.; Ghidina, M.; Bianco, G.; Bernardelli, E.; Pucher, E.; et al. Registro Italiano Pacemaker e Defibrillatori—Bollettino Periodico 2016. Associazione Italiana di Aritmologia e Cardiostimolazione. G. Ital. Cardiol. 2018, 19, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Aktaa, S.; Abdin, A.; Arbelo, E.; Burri, H.; Vernooy, K.; Blomström-Lundqvist, C.; Boriani, G.; Defaye, P.; Deharo, J.-C.; Drossart, I.; et al. European Society of Cardiology Quality Indicators for the care and outcomes of cardiac pacing: Developed by the Working Group for Cardiac Pacing Quality Indicators in collaboration with the European Heart Rhythm Association of the European Society of Cardiology. Europace 2022, 24, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.R.; Zornoff, L.A.M. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq. Bras. Cardiol. 2016, 106, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Wecke, L.; Rubulis, A.; Lundahl, G.; Rosen, M.R.; Bergfeldt, L. Right ventricular pacing–induced electrophysiological remodeling in the human heart and its relationship to cardiac memory. Heart Rhythm 2007, 4, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Luchner, A.; Borgeson, D.D.; Grantham, J.A.; Friedrich, E.; Riegger, G.A.J.; Burnett, J.C.; Redfield, M.M. Relationship between left ventricular wall stress and ANP gene expression during the evolution of rapid ventricular pacing-induced heart failure in the dog. Eur. J. Heart Fail. 2000, 2, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N. Differences in atrial versus ventricular remodeling in dogs with ventricular tachypacing-induced congestive heart failure. Cardiovasc. Res. 2004, 63, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Gavaghan, C. Pacemaker Induced Cardiomyopathy: An Overview of Current Literature. Curr. Cardiol. Rev. 2022, 18, e010921196020. [Google Scholar] [CrossRef]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling—Concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef]
- Koo, A.; Stein, A.; Walsh, R. Pacing-induced Cardiomyopathy. Clin. Pract. Cases Emerg. Med. 2017, 1, 362–364. [Google Scholar] [CrossRef]
- Gould, J.; Sieniewicz, B.; Porter, B.; Sidhu, B.; Rinaldi, C.A. Chronic Right Ventricular Pacing in the Heart Failure Population. Curr. Heart Fail. Rep. 2018, 15, 61–69. [Google Scholar] [CrossRef]
- Mazza, A.; Bendini, M.G.; Leggio, M.; Riva, U.; Ciardiello, C.; Valsecchi, S.; De Cristofaro, R.; Giordano, G. Incidence and predictors of heart failure hospitalization and death in permanent pacemaker patients: A single-centre experience over medium-term follow-up. Europace 2013, 15, 1267–1272. [Google Scholar] [CrossRef]
- Shah, S.; Henry, A.; Roselli, C.; Lin, H.; Sveinbjörnsson, G.; Fatemifar, G.; Hedman, Å.K.; Wilk, J.B.; Morley, M.P.; Chaffin, M.D.; et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat. Commun. 2020, 11, 163. [Google Scholar] [CrossRef]
- Anand, P.; Brown, J.D.; Lin, C.Y.; Qi, J.; Zhang, R.; Artero, P.C.; Alaiti, M.A.; Bullard, J.; Alazem, K.; Margulies, K.B.; et al. BET Bromodomains Mediate Transcriptional Pause Release in Heart Failure. Cell 2013, 154, 569–582. [Google Scholar] [CrossRef]
- Sweeney, M.O.; Hellkamp, A.S.; Ellenbogen, K.A.; Greenspon, A.J.; Freedman, R.A.; Lee, K.L.; Lamas, G.A. Adverse Effect of Ventricular Pacing on Heart Failure and Atrial Fibrillation Among Patients with Normal Baseline QRS Duration in a Clinical Trial of Pacemaker Therapy for Sinus Node Dysfunction. Circulation 2003, 107, 2932–2937. [Google Scholar] [CrossRef]
- Burchfield, J.S.; Xie, M.; Hill, J.A. Pathological Ventricular Remodeling: Mechanisms: Part 1 of 2. Circulation 2013, 128, 388–400. [Google Scholar] [CrossRef]
- Oka, T.; Xu, J.; Molkentin, J.D. Re-employment of developmental transcription factors in adult heart disease. Semin. Cell Dev. Biol. 2007, 18, 117–131. [Google Scholar] [CrossRef]
- Cimiotti, D.; Fujita-Becker, S.; Möhner, D.; Smolina, N.; Budde, H.; Wies, A.; Morgenstern, L.; Gudkova, A.; Sejersen, T.; Sjöberg, G.; et al. Infantile restrictive cardiomyopathy: cTnI-R170G/W impair the interplay of sarcomeric proteins and the integrity of thin filaments. PLoS ONE 2020, 15, e0229227. [Google Scholar] [CrossRef]
- Tucker, N.R.; McLellan, M.A.; Hu, D.; Ye, J.; Parsons, V.A.; Mills, R.W.; Clauss, S.; Dolmatova, E.; Shea, M.A.; Milan, D.J.; et al. Novel Mutation in FLNC (Filamin C) Causes Familial Restrictive Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001780. [Google Scholar] [CrossRef]
- Kiselev, A.; Vaz, R.; Knyazeva, A.; Khudiakov, A.; Tarnovskaya, S.; Liu, J.; Sergushichev, A.; Kazakov, S.; Frishman, D.; Smolina, N.; et al. De novo mutations in FLNC leading to early-onset restrictive cardiomyopathy and congenital myopathy. Hum. Mutat. 2018, 39, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Hain, C.; Flottmann, F.; Ratnavadivel, S.; Gaertner, A.; Klauke, B.; Kalinowski, J.; Körperich, H.; Gummert, J.; Paluszkiewicz, L.; et al. The Desmin mutation DES-c.735G>C causes severe restrictive cardiomyopathy by inducing in-frame skipping of exon-3. Biomedicines 2021, 9, 1400. [Google Scholar] [CrossRef] [PubMed]
- Park, R.C.; Little, W.C.; O’Rourke, R.A. Effect of alteration of left ventricular activation sequence on the left ventricular end-systolic pressure-volume relation in closed-chest dogs. Circ. Res. 1985, 57, 706–717. [Google Scholar] [CrossRef]
- Veltrop, R.J.A.; Kukk, M.M.; Topouzidou, K.; Didden, L.; Muchir, A.; Van Steenbeek, F.G.; Schurgers, L.J.; Harakalova, M. From gene to mechanics: A comprehensive insight into the mechanobiology of LMNA mutations in cardiomyopathy. Cell Commun. Signal. 2024, 22, 197. [Google Scholar] [CrossRef] [PubMed]
- Cheedipudi, S.M.; Matkovich, S.J.; Coarfa, C.; Hu, X.; Robertson, M.J.; Sweet, M.; Taylor, M.; Mestroni, L.; Cleveland, J.; Willerson, J.T.; et al. Genomic Reorganization of Lamin-Associated Domains in Cardiac Myocytes Is Associated with Differential Gene Expression and DNA Methylation in Human Dilated Cardiomyopathy. Circ. Res. 2019, 124, 1198–1213. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.; Frese, K.S.; Park, Y.J.; Keller, A.; Vogel, B.; Lindroth, A.M.; Weichenhan, D.; Franke, J.; Fischer, S.; Bauer, A.; et al. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol. Med. 2013, 5, 413–429. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Garrido, M.; Chapski, D.J.; Schmitt, A.D.; Kimball, T.H.; Karbassi, E.; Monte, E.; Balderas, E.; Pellegrini, M.; Shih, T.-T.; Soehalim, E.; et al. High-Resolution Mapping of Chromatin Conformation in Cardiac Myocytes Reveals Structural Remodeling of the Epigenome in Heart Failure. Circulation 2017, 136, 1613–1625. [Google Scholar] [CrossRef] [PubMed]
- Simantirakis, Ε.; Arkolaki, E.; Kontaraki, J.; Chlouverakis, G.; Mavrakis, H.; Kallergis, E.; Parthenakis, F.; Vardas, P. The impact of paced QRS duration on the expression of genes related to contractile function of the left ventricle in chronically paced patients from the right ventricular apex. Hellenic J. Cardiol. 2020, 61, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Arkolaki, E.G.; Simantirakis, E.N.; Kontaraki, J.E.; Chrysostomakis, S.I.; Patrianakos, A.P.; Chlouverakis, G.I.; Nakou, E.S.; Vardas, P.E. Alterations in the expression of genes related to contractile function and hypertrophy of the left ventricle in chronically paced patients from the right ventricular apex. Europace 2015, 17, 1563–1570. [Google Scholar] [CrossRef]
- Shareef, M.A.; Anwer, L.A.; Poizat, C. Cardiac SERCA2A/B: Therapeutic targets for heart failure. Eur. J. Pharmacol. 2014, 724, 1–8. [Google Scholar] [CrossRef]
- Hayward, C.; Banner, N.R.; Morley-Smith, A.; Lyon, A.R.; Harding, S.E. The Current and Future Landscape of SERCA Gene Therapy for Heart Failure: A Clinical Perspective. Hum. Gene Ther. 2015, 26, 293–304. [Google Scholar] [CrossRef]
- Lowes, B.D.; Gilbert, E.M.; Abraham, W.T.; Minobe, W.A.; Larrabee, P.; Ferguson, D.; Wolfel, E.E.; Lindenfeld, J.; Tsvetkova, T.; Robertson, A.D.; et al. Myocardial Gene Expression in Dilated Cardiomyopathy Treated with Beta-Blocking Agents. N. Engl. J. Med. 2002, 346, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Vanderheyden, M.; Mullens, W.; Delrue, L.; Goethals, M.; De Bruyne, B.; Wijns, W.; Geelen, P.; Verstreken, S.; Wellens, F.; Bartunek, J. Myocardial Gene Expression in Heart Failure Patients Treated with Cardiac Resynchronization Therapy. J. Am. Coll. Cardiol. 2008, 51, 129–136. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003, 4, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.-C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.W.; et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Eijgenraam, T.R.; Boukens, B.J.; Boogerd, C.J.; Schouten, E.M.; Van De Kolk, C.W.A.; Stege, N.M.; Te Rijdt, W.P.; Hoorntje, E.T.; Van Der Zwaag, P.A.; Van Rooij, E.; et al. The phospholamban p.(Arg14del) pathogenic variant leads to cardiomyopathy with heart failure and is unresponsive to standard heart failure therapy. Sci. Rep. 2020, 10, 9819. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Xie, X.; Li, J.; Zhang, Y.; Xu, C.; Yang, J. Early Right Ventricular Apical Pacing-Induced Gene Expression Alterations Are Associated with Deterioration of Left Ventricular Systolic Function. Dis. Markers 2017, 2017, 8405196. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; Sen, S.; Vela, D. Return to the fetal gene program: A suggested metabolic link to gene expression in the heart. Ann. N. Y. Acad. Sci. 2010, 1188, 191–198. [Google Scholar] [CrossRef]
- Leri, A.; Liu, Y.; Malhotra, A.; Li, Q.; Stiegler, P.; Claudio, P.P.; Giordano, A.; Kajstura, J.; Hintze, T.H.; Anversa, P. Pacing-Induced Heart Failure in Dogs Enhances the Expression of p53 and p53-Dependent Genes in Ventricular Myocytes. Circulation 1998, 97, 194–203. [Google Scholar] [CrossRef]
- Haider, N.; Narula, N.; Narula, J. Apoptosis in heart failure represents programmed cell survival, not death, of cardiomyocytes and likelihood of reverse remodeling. J. Card. Fail. 2002, 8, S512–S517. [Google Scholar] [CrossRef]
- Young, M.E.; McNulty, P.; Taegtmeyer, H. Adaptation and Maladaptation of the Heart in Diabetes: Part II: Potential Mechanisms. Circulation 2002, 105, 1861–1870. [Google Scholar] [CrossRef]
- Wu, Y.; Bell, S.P.; Trombitas, K.; Witt, C.C.; Labeit, S.; LeWinter, M.M.; Granzier, H. Changes in Titin Isoform Expression in Pacing-Induced Cardiac Failure Give Rise to Increased Passive Muscle Stiffness. Circulation 2002, 106, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, M.; Kassiotis, C.; Razeghi, P.; Taegtmeyer, H. Return to the fetal gene program protects the stressed heart: A strong hypothesis. Heart Fail. Rev. 2007, 12, 331–343. [Google Scholar] [CrossRef]
- Akazawa, H.; Komuro, I. Roles of Cardiac Transcription Factors in Cardiac Hypertrophy. Circ. Res. 2003, 92, 1079–1088. [Google Scholar] [CrossRef]
- Kerkelä, R.; Pikkarainen, S.; Majalahti-Palviainen, T.; Tokola, H.; Ruskoaho, H. Distinct Roles of Mitogen-activated Protein Kinase Pathways in GATA-4 Transcription Factor-mediated Regulation of B-type Natriuretic Peptide Gene. J. Biol. Chem. 2002, 277, 13752–13760. [Google Scholar] [CrossRef]
- Barth, A.S.; Aiba, T.; Halperin, V.; DiSilvestre, D.; Chakir, K.; Colantuoni, C.; Tunin, R.S.; Dimaano, V.L.; Yu, W.; Abraham, T.P.; et al. Cardiac Resynchronization Therapy Corrects Dyssynchrony-Induced Regional Gene Expression Changes on a Genomic Level. Circ. Cardiovasc. Genet. 2009, 2, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J.; Goldenthal, M.J.; Damle, S.; Pi, Y.; Moe, G.W. Regional Distribution of Mitochondrial Dysfunction and Apoptotic Remodeling in Pacing-Induced Heart Failure. J. Card. Fail. 2009, 15, 700–708. [Google Scholar] [CrossRef]
- Mahmood, A.; Ahmed, K.; Zhang, Y. β-Adrenergic Receptor Desensitization/Down-Regulation in Heart Failure: A Friend or Foe? Front. Cardiovasc. Med. 2022, 9, 925692. [Google Scholar] [CrossRef]
- Al-Hesayen, A.; Parker, J.D. Adverse effects of atrioventricular synchronous right ventricular pacing on left ventricular sympathetic activity, efficiency, and hemodynamic status. Am. J. Physiol.-Heart Circ. Physiol. 2006, 291, H2377–H2379. [Google Scholar] [CrossRef]
- Thames, M.D. Effect of d- and l-propranolol on the discharge of cardiac vagal C fibers. Am. J. Physiol.-Heart Circ. Physiol. 1980, 238, H465–H470. [Google Scholar] [CrossRef]
- Hamdan, M.H.; Zagrodzky, J.D.; Joglar, J.A.; Sheehan, C.J.; Ramaswamy, K.; Erdner, J.F.; Page, R.L.; Smith, M.L. Biventricular Pacing Decreases Sympathetic Activity Compared with Right Ventricular Pacing in Patients with Depressed Ejection Fraction. Circulation 2000, 102, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Saxon, L.A.; Kerwin, W.F.; Cahalan, M.K.; Kalman, J.M.; Olgin, J.E.; Foster, E.; Schiller, N.B.; Shinbane, J.S.; Lesh, M.D.; Merrick, S.H. Acute Effects of Intraoperative Multisite Ventricular Pacing on Left Ventricular Function and Activation/Contraction Sequence in Patients with Depressed Ventricular Function. J. Cardiovasc. Electrophysiol. 1998, 9, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Luchner, A.; Stevens, T.L.; Borgeson, D.D.; Redfield, M.M.; Bailey, J.E.; Sandberg, S.M.; Heublein, D.M.; Burnett, J.C. Angiotensin II in the Evolution of Experimental Heart Failure. Hypertension 1996, 28, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Schnee, J. Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc. Res. 2000, 46, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Agha, M.; Rikhi, R.; Hari, K.; Bodziock, G.; Bhave, P. Abstract 17657: The Effect of Medical Therapy on Reducing the Risk of Pacing Induced Cardiomyopathy. Circulation 2023, 148, 1001S–1067S. [Google Scholar] [CrossRef]
- Kajstura, J.; Zhang, X.; Liu, Y.; Szoke, E.; Cheng, W.; Olivetti, G.; Hintze, T.H.; Anversa, P. The Cellular Basis of Pacing-Induced Dilated Cardiomyopathy: Myocyte Cell Loss and Myocyte Cellular Reactive Hypertrophy. Circulation 1995, 92, 2306–2317. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.M.; Yu, Z.X.; Ferrans, V.J.; Lowenstein, R.A.; Finkel, T. Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93, 11848–11852. [Google Scholar] [CrossRef]
- Naspro, R.; Rossini, R.; Musumeci, G.; Gadda, F.; Pozzo, L.F.D. Antiplatelet Therapy in Patients with Coronary Stent Undergoing Urologic Surgery: Is It Still No Man’s Land? Eur. Urol. 2013, 64, 101–105. [Google Scholar] [CrossRef]
- Sepúlveda, M.; Gonano, L.A.; Back, T.G.; Chen, S.R.W.; Vila Petroff, M. Role of CaMKII and ROS in rapid pacing-induced apoptosis. J. Mol. Cell. Cardiol. 2013, 63, 135–145. [Google Scholar] [CrossRef]
- Klug, D.; Boule, S.; Wissocque, L.; Montaigne, D.; Marechal, X.; Hassoun, S.M.; Neviere, R. Right Ventricular Pacing with Mechanical Dyssynchrony Causes Apoptosis Interruptus and Calcium Mishandling. Can. J. Cardiol. 2013, 29, 510–518. [Google Scholar] [CrossRef]
- Marín-García, J. Abnormal cardiac and skeletal muscle mitochondrial function in pacing-induced cardiac failure. Cardiovasc. Res. 2001, 52, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Passino, C.; Barison, A.; Vergaro, G.; Gabutti, A.; Borrelli, C.; Emdin, M.; Clerico, A. Markers of fibrosis, inflammation, and remodeling pathways in heart failure. Clin. Chim. Acta 2015, 443, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, P.W.; Moe, G.W.; Howard, R.J.; Grima, E.A.; Cruz, T.F. Structural remodelling in heart failure: Gelatinase induction. Can. J. Cardiol. 1994, 10, 214–220. [Google Scholar] [PubMed]
- Lin, J.; Lai, L.; Lin, C.; Chou, N.; Chiu, C.; Lin, J. Left Ventricular Extracellular Matrix Remodeling in Dogs with Right Ventricular Apical Pacing. J. Cardiovasc. Electrophysiol. 2010, 21, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, G.F. Pacing-Induced Remodeling of the Ventricle: Fire in the Matrix. J. Cardiovasc. Electrophysiol. 2010, 21, 1150–1152. [Google Scholar] [CrossRef] [PubMed]
- Senzaki, H.; Paolocci, N.; Gluzband, Y.A.; Lindsey, M.L.; Janicki, J.S.; Crow, M.T.; Kass, D.A. β-Blockade Prevents Sustained Metalloproteinase Activation and Diastolic Stiffening Induced by Angiotensin II Combined with Evolving Cardiac Dysfunction. Circ. Res. 2000, 86, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Hoit, B.D.; Takeishi, Y.; Cox, M.J.; Gabel, M.; Kirkpatrick, D.; Walsh, R.A.; Tyagi, S.C. Remodeling of the left atrium in pacing-induced atrial cardiomyopathy. Mol. Cell. Biochem. 2002, 238, 145–150. [Google Scholar] [CrossRef]
- Birner, C.M.; Ulucan, C.; Fredersdorf, S.; Rihm, M.; Löwel, H.; Stritzke, J.; Schunkert, H.; Hengstenberg, C.; Holmer, S.; Riegger, G.; et al. Head-to-head comparison of BNP and IL-6 as markers of clinical and experimental heart failure: Superiority of BNP. Cytokine 2007, 40, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Yuan, Y.; Li, J.; Gionfriddo, M.R.; Huang, R. Tumor necrosis factor-α and its role as a mediator in myocardial infarction: A brief review. Chronic Dis. Transl. Med. 2015, 1, 18–26. [Google Scholar] [CrossRef]
- Châtelain, P.; Adamec, R.; Cox, J.N. Morphological changes in human myocardium during permanent pacing. Virchows Arch. A Pathol. Anat. Histopathol. 1985, 407, 43–57. [Google Scholar] [CrossRef]
- Yamazaki, K.G.; Villarreal, F.J. Ventricular pacing-induced loss of contractile function and development of epicardial inflammation. Am. J. Physiol.-Heart Circ. Physiol. 2011, 300, H1282–H1290. [Google Scholar] [CrossRef] [PubMed]
- Kirk, J.A.; Kass, D.A. Electromechanical dyssynchrony and resynchronization of the failing heart. Circ. Res. 2013, 113, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, J.; Yuzyuk, T.N.; Cox, J.; Makaju, A.; Miller, M.; Lichter, J.; Li, H.; Leavy, J.D.; Franklin, S.; Zaitsev, A.V. Metabolic remodeling in moderate synchronous versus dyssynchronous pacing-induced heart failure: Integrated metabolomics and proteomics study. PLoS ONE 2015, 10, e0118974. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.-M.; Dzeja, P.P.; Shen, W.K.; Jahangir, A.; Hart, C.Y.T.; Terzic, A.; Redfield, M.M. Failing atrial myocardium: Energetic deficits accompany structural remodeling and electrical instability. Am. J. Physiol.-Heart Circ. Physiol. 2003, 284, H1313–H1320. [Google Scholar] [CrossRef] [PubMed]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Utsumi, H.; Kang, D.; Hattori, N.; Uchida, K.; Arimura, K.; Egashira, K.; Takeshita, A. Mitochondrial Electron Transport Complex I Is a Potential Source of Oxygen Free Radicals in the Failing Myocardium. Circ. Res. 1999, 85, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Osorio, J.C.; Stanley, W.C.; Linke, A.; Castellari, M.; Diep, Q.N.; Panchal, A.R.; Hintze, T.H.; Lopaschuk, G.D.; Recchia, F.A. Impaired Myocardial Fatty Acid Oxidation and Reduced Protein Expression of Retinoid X Receptor-α in Pacing-Induced Heart Failure. Circulation 2002, 106, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Prinzen, F.W.; Peschar, M. Relation Between the Pacing Induced Sequence of Activation and Left Ventricular Pump Function in Animals. Pacing Clin. Electrophysiol. 2002, 25, 484–498. [Google Scholar] [CrossRef] [PubMed]
- López, B.; Ravassa, S.; Moreno, M.U.; José, G.S.; Beaumont, J.; González, A.; Díez, J. Diffuse myocardial fibrosis: Mechanisms, diagnosis and therapeutic approaches. Nat. Rev. Cardiol. 2021, 18, 479–498. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.R.; MacLeod, K.T.; Zhang, Y.; Garcia, E.; Kanda, G.K.; Lab, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc. Natl. Acad. Sci. USA 2009, 106, 6854–6859. [Google Scholar] [CrossRef]
- Khurwolah, M.R.; Yao, J.; Kong, X.-Q. Adverse Consequences of Right Ventricular Apical Pacing and Novel Strategies to Optimize Left Ventricular Systolic and Diastolic Function. Curr. Cardiol. Rev. 2019, 15, 145–155. [Google Scholar] [CrossRef]
- Barold, S.S.; Ovsyshcher, I.E. Pacemaker-Induced Mitral Regurgitation. Pacing Clin. Electrophysiol. 2005, 28, 357–360. [Google Scholar] [CrossRef]
- Van Oosterhout, M.F.M.; Prinzen, F.W.; Arts, T.; Schreuder, J.J.; Vanagt, W.Y.R.; Cleutjens, J.P.M.; Reneman, R.S. Asynchronous Electrical Activation Induces Asymmetrical Hypertrophy of the Left Ventricular Wall. Circulation 1998, 98, 588–595. [Google Scholar] [CrossRef]
- Blaauw, E.; Lorenzen-Schmidt, I.; Babiker, F.A.; Munts, C.; Prinzen, F.W.; Snoeckx, L.H.; Van Bilsen, M.; Van Der Vusse, G.J.; Van Nieuwenhoven, F.A. Stretch-Induced Upregulation of Connective Tissue Growth Factor in Rabbit Cardiomyocytes. J Cardiovasc. Transl. Res. 2013, 6, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Herum, K.M.; Choppe, J.; Kumar, A.; Engler, A.J.; McCulloch, A.D. Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol. Biol. Cell 2017, 28, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Badke, F.R.; Boinay, P.; Covell, J.W. Effects of ventricular pacing on regional left ventricular performance in the dog. Am. J. Physiol.-Heart Circ. Physiol. 1980, 238, H858–H867. [Google Scholar] [CrossRef]
- Matsuoka, K.; Nishino, M.; Kato, H.; Egami, Y.; Shutta, R.; Yamaguchi, H.; Tanaka, K.; Tanouchi, J.; Yamada, Y. Right Ventricular Apical Pacing Impairs Left Ventricular Twist as Well as Synchrony: Acute Effects of Right Ventricular Apical Pacing. J. Am. Soc. Echocardiogr. 2009, 22, 914–919. [Google Scholar] [CrossRef]
- Burkhoff, D.; Oikawa, R.Y.; Sagawa, K. Influence of pacing site on canine left ventricular contraction. Am. J. Physiol.-Heart Circ. Physiol. 1986, 251, H428–H435. [Google Scholar] [CrossRef]
- Gauthey, A.; Willemen, E.; Lumens, J.; Ploux, S.; Bordachar, P.; Ritter, P.; Prinzen, F.W.; Lejeune, S.; Pouleur, A.; Garnir, Q.; et al. Impact of paced left ventricular dyssynchrony on left ventricular reverse remodeling after cardiac resynchronization therapy. J. Cardiovasc. Electrophysiol. 2020, 31, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Sachse, F.B.; Torres, N.S.; Savio-Galimberti, E.; Aiba, T.; Kass, D.A.; Tomaselli, G.F.; Bridge, J.H. Subcellular Structures and Function of Myocytes Impaired During Heart Failure Are Restored by Cardiac Resynchronization Therapy. Circ. Res. 2012, 110, 588–597. [Google Scholar] [CrossRef]
- Kiehl, E.L.; Makki, T.; Kumar, R.; Gumber, D.; Kwon, D.H.; Rickard, J.W.; Kanj, M.; Wazni, O.M.; Saliba, W.I.; Varma, N.; et al. Incidence and predictors of right ventricular pacing-induced cardiomyopathy in patients with complete atrioventricular block and preserved left ventricular systolic function. Heart Rhythm 2016, 13, 2272–2278. [Google Scholar] [CrossRef]
- Kaye, G.; Ng, J.Y.; Ahmed, S.; Valencia, D.; Harrop, D.; Ng, A.C.T. The Prevalence of Pacing-Induced Cardiomyopathy (PICM) in Patients with Long Term Right Ventricular Pacing—Is it a Matter of Definition? Heart Lung Circ. 2019, 28, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Parakh, N.; Gupta, A.; Juneja, R.; Naik, N.; Yadav, R.; Sharma, G.; Roy, A.; Verma, S.K.; Bahl, V.K. Incidence and predictors of pacemaker-induced cardiomyopathy with comparison between apical and non-apical right ventricular pacing sites. J. Interv. Card. Electrophysiol. 2019, 56, 63–70. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Study Group | Gene Expression | Follow-Up | Findings | |
|---|---|---|---|---|
| Luchner et al. [7] | Eleven dogs underwent implantation of a programmable cardiac pacemaker for rapid ventricular pacing. | NPPA gene expression levels were evaluated in myocardial tissue samples. | 38 days | NPPA gene levels were elevated in all samples from overt CHF ventricular tissue samples compared to being barely detectable in control and asymptomatic left ventricular dysfunction dogs. A positive signal for NPPA mRNA was present in all left atrial samples from the control, asymptomatic left ventricular dysfunction, and overt CHF animals. Left atrial ANP concentrations exceeded left ventricular ANP and showed a tendency to increase during overt CHF. |
| Arkolaki et al. [28] | 52 patients with bradycardic conditions (group A-24 individuals AVCD and group B-28 patients with sinus node disease). | SERCA2, MYH6, and MYH7 gene expression levels were evaluated in peripheral blood. | 12 months | In group A, SERCA2 and MYH6 genes decreased compared to MYH7 which increased. In total, 25% of patients from group A demonstrated significant LV remodeling. Early alterations in gene expression were associated with a deterioration in LV function and geometry that became apparent months later on echocardiographic evaluation. |
| Vanderheyden et al. [33] | Twenty-four patients were referred for CRT implantation. | MYH6, MYH7, and NPPB gene expression levels were evaluated in the myocardial tissue sample. | 4 months | Responders to CRT were associated with an increase in MYH6 mRNA levels and the ratio of α-/β-MHC proteins. Furthermore, a decrease in NPPB mRNA levels was observed in the same population. |
| Eijgenraam et al. [36] | 6 wild-type mice, 10 heterozygous (PLNR14Δ/+) mice and 13 homozygous (PLN-R14 Δ/Δ) mice | Homozygous mice showed upregulation of fetal genes as follows: elevated expression of the NPPA and NPPB gene and increase in the ratio of MYH7/MYH6 gene expression | 20 months | Mice PLN-R14 Δ/Δ demonstrated increased LV end-diastolic and end-systolic volumes, decreased stroke volume and ejection fraction, as well as significantly decreased survival compared to wild-type mice Elevated LV expression of fibrotic genes was observed in homozygous mice Heterozygous mice developed cardiomyopathy at a later age; cardiac sections showed PLN protein aggregation in some of the cardiomyocytes and an increase in myocardial fibrosis |
| Xu et al. [37] | 60 patients with complete AV block and preserved LVEF (≥50%) (group A-30 RVA pacing, group B-RVOT pacing). | OPA1 and SERCA2a gene expression levels were evaluated in peripheral blood. | 24 months | OPA1 and SERCA2a mRNA levels were significantly lower in group A compared to group B. Compared with the baseline value, the mRNA levels of SERCA2a and OPA1 were decreased in group A in the first month of evaluation. In group B, this difference was not statistically significant between the initial and final mRNA levels of both genes. |
| Wu et al. [42] | Six tachycardia-induced dilated cardiomyopathy canine models compared to 14 controls. | N2B titin isoform (short spring), N2BA titin isoform (long spring), and obscurin- 800 kDa elastic protein were involved in signaling processes in sarcomeric restructuring and were evaluated in myocardial tissue samples. | 4 weeks | In paced animals, in contrast to the control, the expression of N2BA titin was reduced and the N2B expression was more prominent. Pacing did not influence the total amount of titin, but the expression level of the stiffer N2B isoform was influenced. Obscurin was upregulated by pacing. |
| Marin-Garcia et al. [47] | Eleven dogs underwent continuous rapid right ventricular pacing compared to ten normal dogs who served as controls. | Large-scale mtDNA deletions were evaluated in myocardial tissue samples. | 3 weeks | Large-scale mtDNA deletion was found to be more likely present in myocardial tissue of paced compared to control animals. |
| Structural Remodeling |
| Microscopic |
| Diffuse fibrosis of ECV |
| Loss of the t-system and separation of RyRs from sarcolemmal structures |
| Macroscopic |
| Asymmetrical LV hypertrophy and dilatation |
| LA dilation |
| TVR |
| Functional Remodeling |
| Reduced stroke volume—Impaired LV twist |
| Decrease in LV filling duration |
| fMR |
| Myocardial coronary perfusion defects |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malikides, O.; Simantirakis, E.; Zacharis, E.; Fragkiadakis, K.; Kochiadakis, G.; Marketou, M. Cardiac Remodeling and Ventricular Pacing: From Genes to Mechanics. Genes 2024, 15, 671. https://doi.org/10.3390/genes15060671
Malikides O, Simantirakis E, Zacharis E, Fragkiadakis K, Kochiadakis G, Marketou M. Cardiac Remodeling and Ventricular Pacing: From Genes to Mechanics. Genes. 2024; 15(6):671. https://doi.org/10.3390/genes15060671
Chicago/Turabian StyleMalikides, Onoufrios, Emmanouel Simantirakis, Evangelos Zacharis, Konstantinos Fragkiadakis, George Kochiadakis, and Maria Marketou. 2024. "Cardiac Remodeling and Ventricular Pacing: From Genes to Mechanics" Genes 15, no. 6: 671. https://doi.org/10.3390/genes15060671