
Gonadal Transcriptome Sequencing Analysis Reveals the Candidate Sex-Related Genes and Signaling Pathways in the East Asian Common Octopus, Octopus sinensis
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Octopus
2.2. Histological Analysis and Samples Collection
2.3. RNA Library Construction, Sequencing and Reference-Based Assembly
2.4. Gene-Expression Analysis and Sample Relationship Analysis
2.5. DEGs Identification and Function Enrichment
2.6. Real-Time Quantitative PCR (RT-qPCR) Verification
3. Results
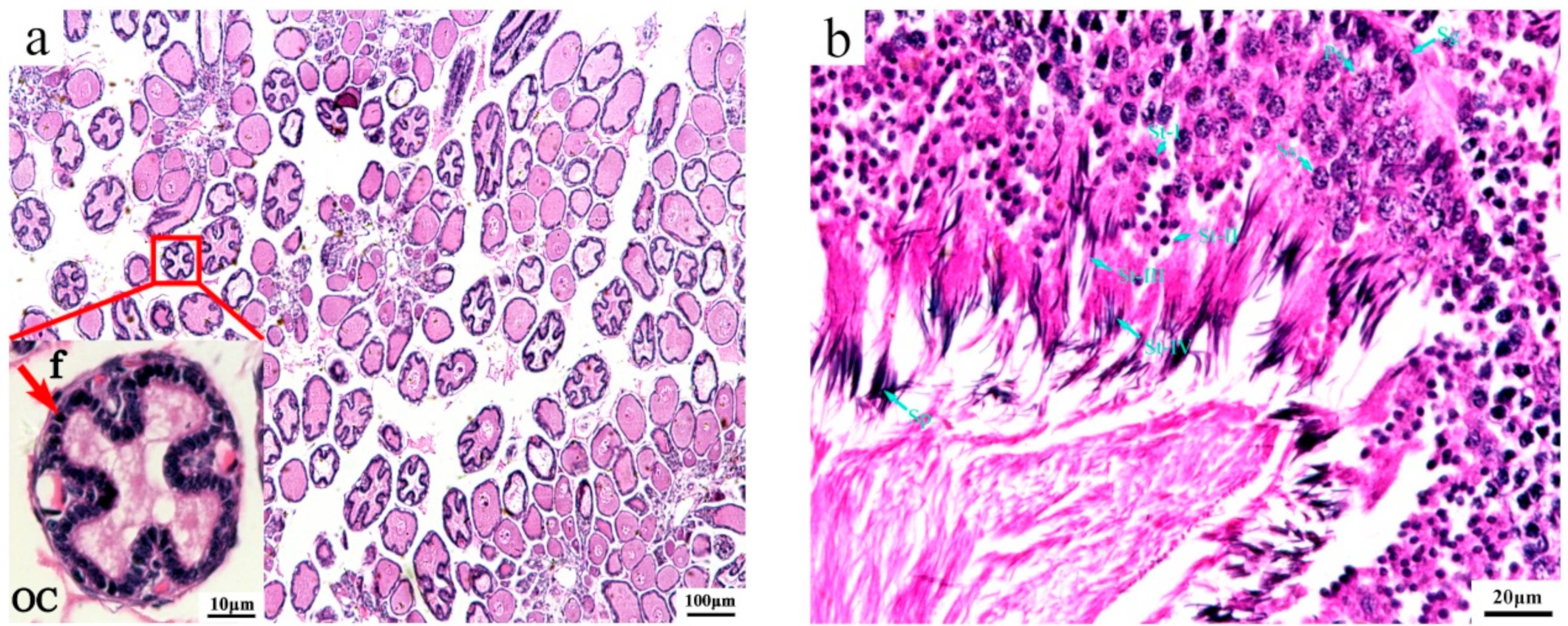
3.1. Histological Structure of Immature Gonads
3.2. Overall Transcriptome and Sequencing Data
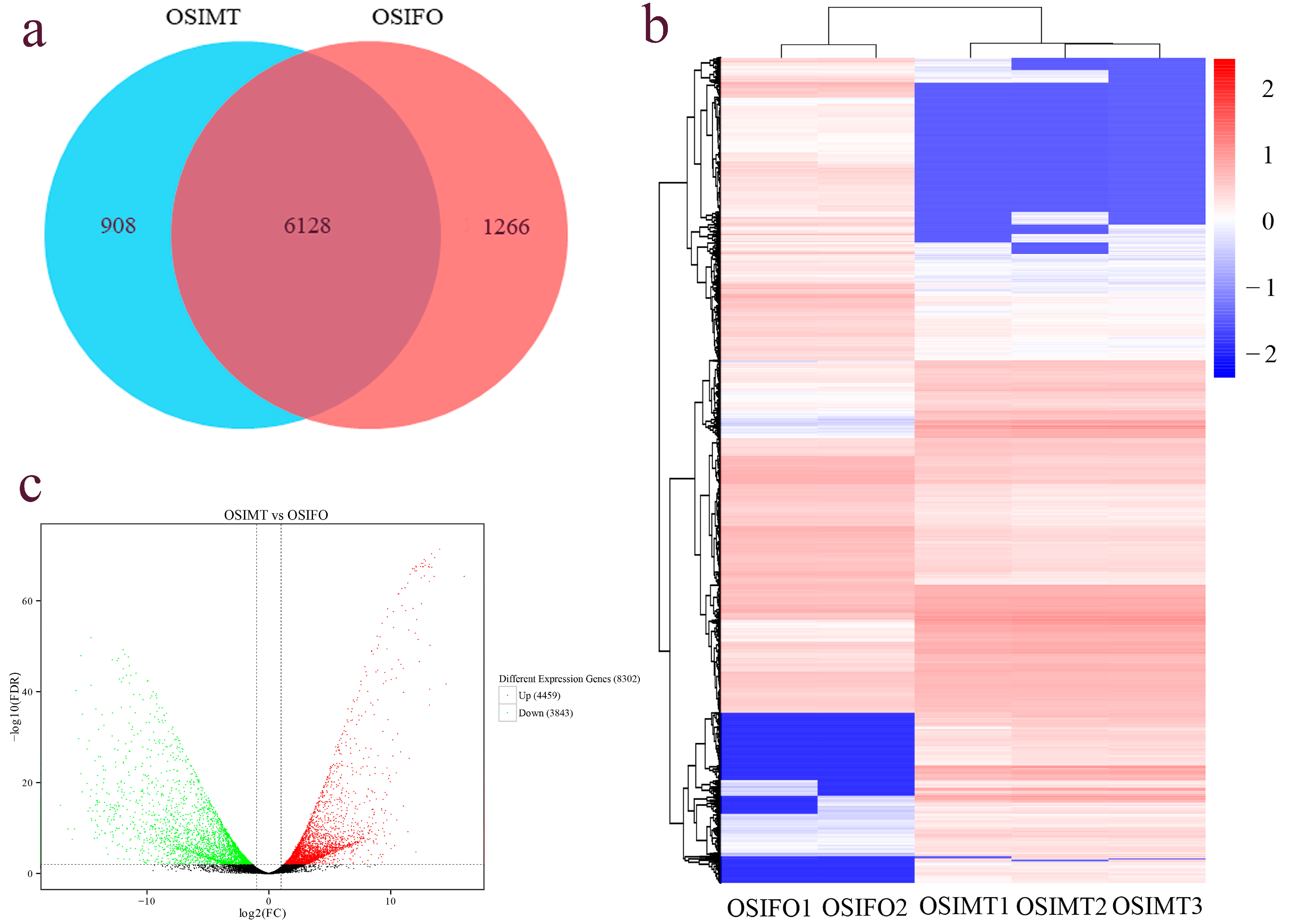
3.3. Different Expression Gene Identification
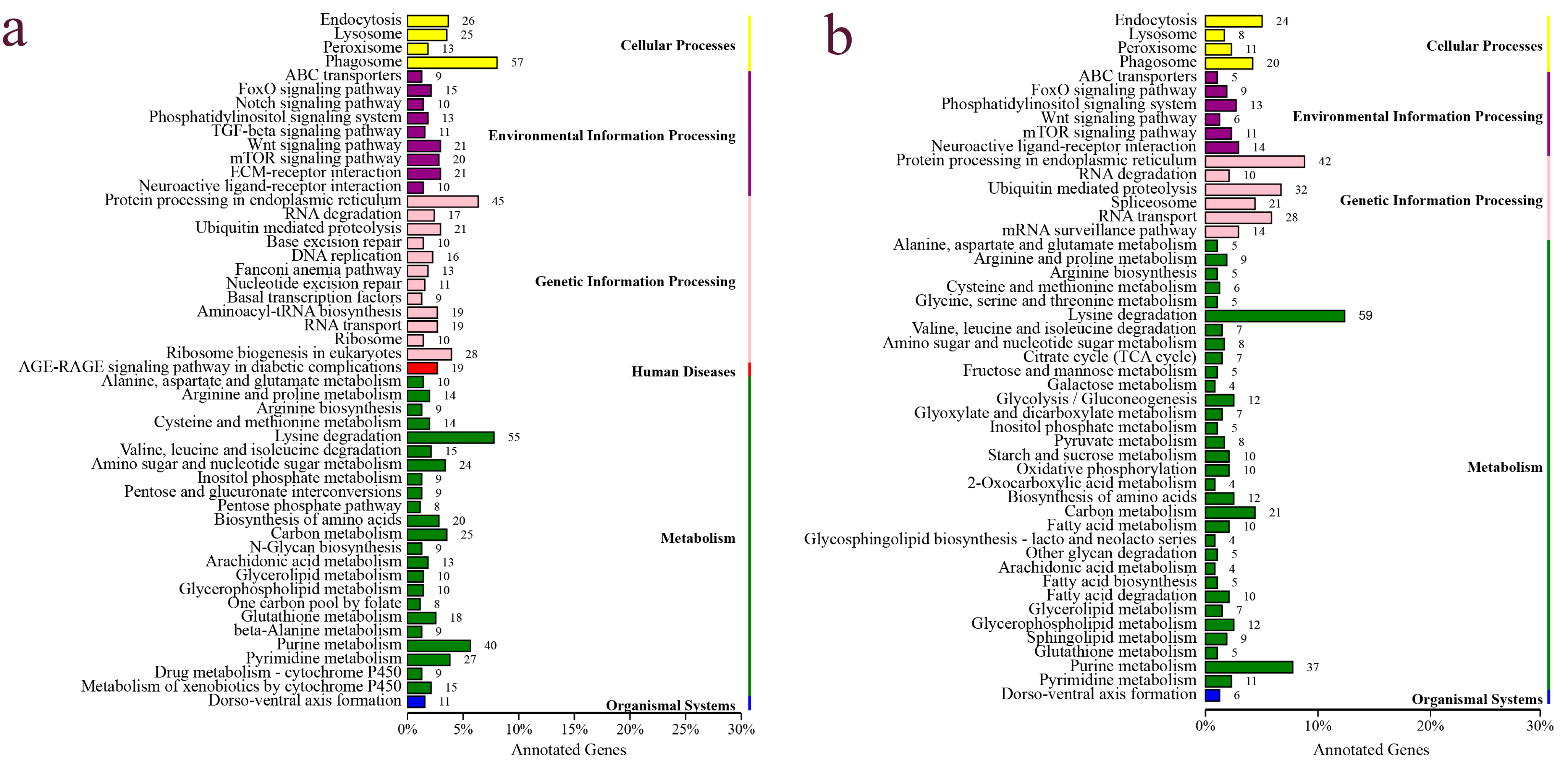
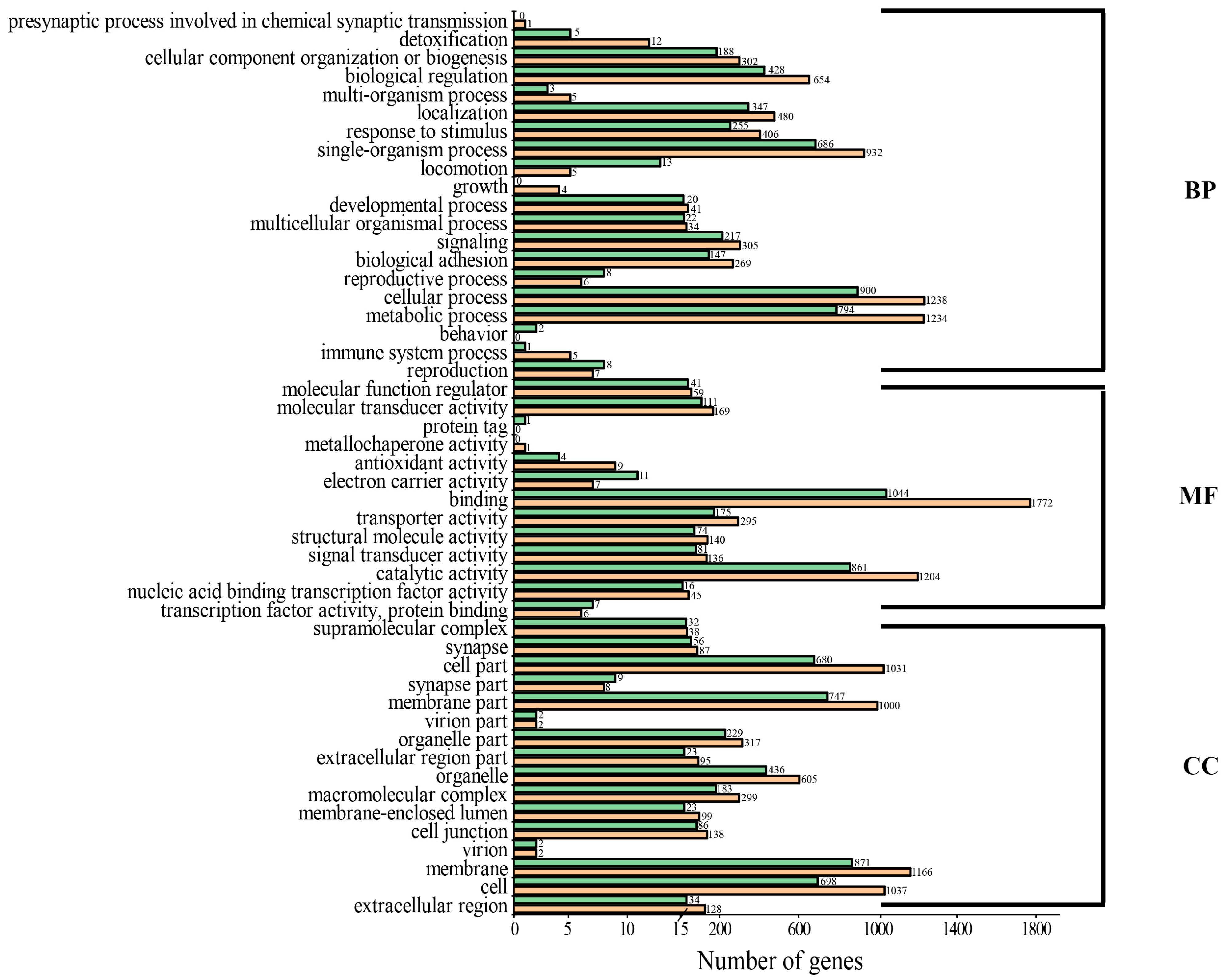
3.4. Functional Annotation, Classification and Enrichment Analysis of DEG
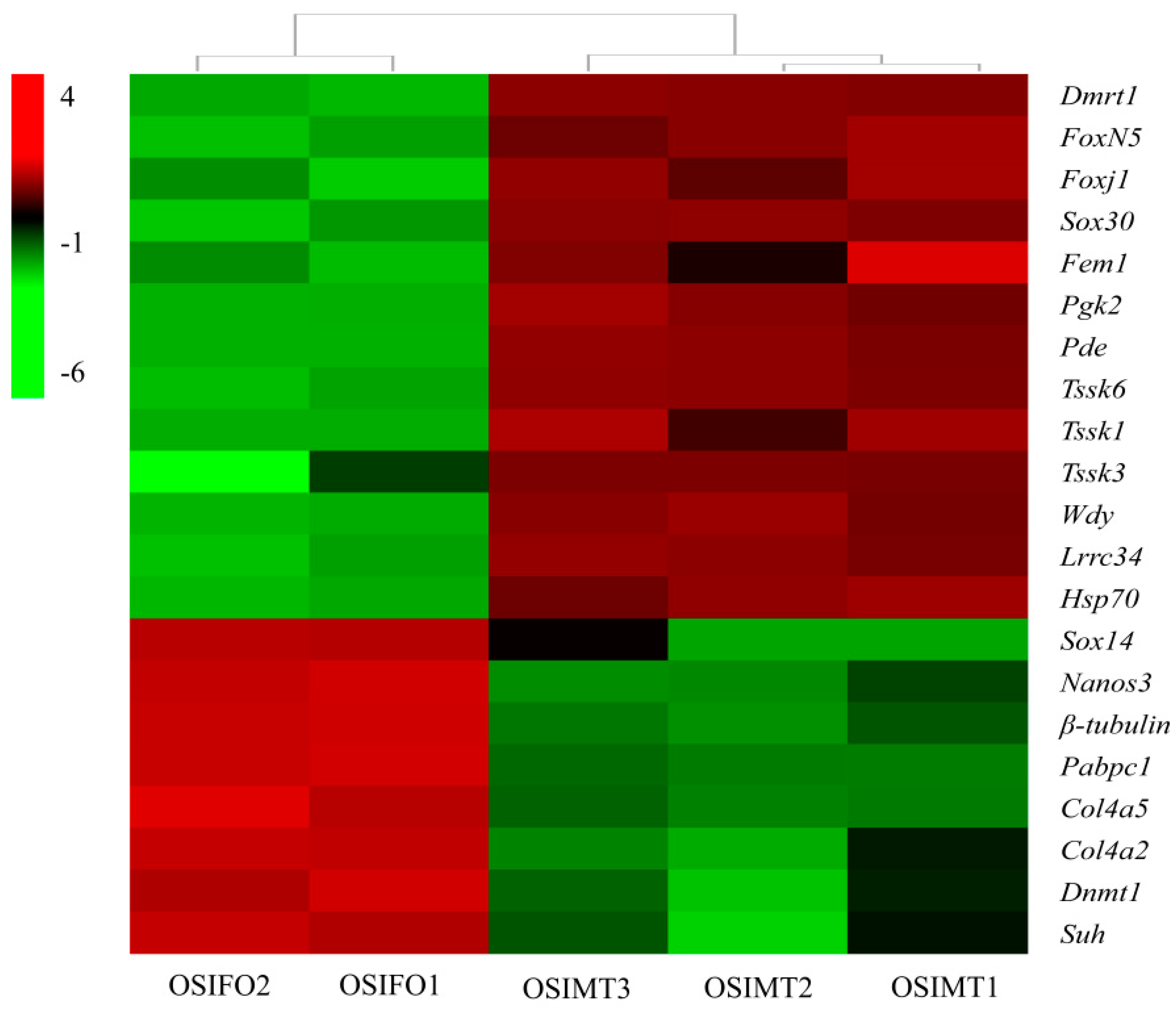
3.5. Identification of Sex-Related Genes in O. sinensis
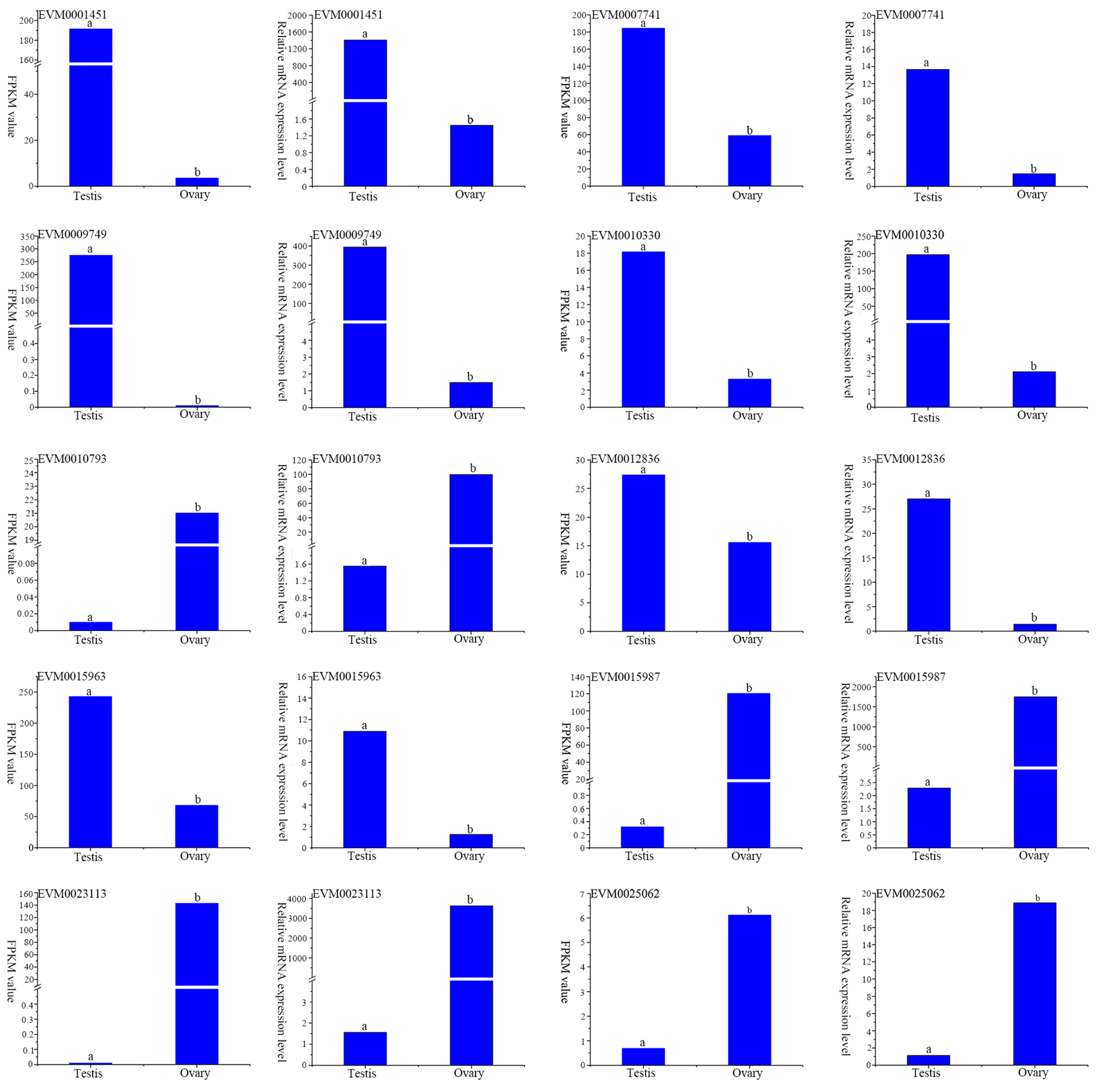
3.6. Real-Time Quantitative PCR (RT-qPCR) Verification
4. Discussion
4.1. Overall Characteristics of the Transcriptome Data
4.2. Signaling Pathways and GO Terms Related to Sex-Determination/differentiation Process
4.3. Key Sex-Related Differentially Expressed Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Capel, B. Vertebrate sex determination: Evolutionary plasticity of a fundamental switch. Nat. Rev. Genet. 2017, 18, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Gui, J.F. Diverse and variable sex determination mechanisms in vertebrates. Sci. China Life Sci. 2018, 61, 1503–1514. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Mei, J.; Ge, C.T.; Liu, X.L.; Gui, J.F. Sex determination mechanisms and sex control approaches in aquaculture animals. Sci. China Life Sci. 2022, 65, 1091–1122. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, Y.; Chakraborty, T.; Paul-Prasanth, B.; Ohta, K.; Nakamura, M. Sex Determination, Gonadal Sex Differentiation and Plasticity in Vertebrate Species. Physiol. Rev. 2021, 101, 1237–1308. [Google Scholar] [CrossRef] [PubMed]
- Verhulst, E.C.; Zande, L.V.D.; Beukeboom, L.W. Insect sex determination: It all evolves around transformer. Curr. Opin. Genet. Dev. 2010, 20, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Wanninger, A.; Wollesen, T. Mollusca. In Evolutionary Developmental Biology of Invertebrates 2: Lophotrochozoa (Spiralia), 1st ed.; Wanninger, A., Ed.; Springer: Vienna, Austria, 2015; Volume VII, pp. 173–176. [Google Scholar]
- Soyez, C.; Huvet, A.; Gueguen, Y.; Lo, C.; Moullac, G.L. Determination of gender in the Pearl oyster Pinctada Margaritifera. J. Shellfish Res. 2011, 30, 231–240. [Google Scholar]
- Santerre, C.; Sourdaine, P.; Mingant, C.; Robert, R.; Martinez, A.S. Oyster sex determination is influenced by temperature—First clues in spat during first gonadic differentiation and gametogenesis. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2013, 165, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Breton, S.; Capt, C.; Guerra, D.; Stewart, D. Sex-determining mechanisms in bivalves. In Transitions between Sexual Systems, 1st ed.; Leonard, J.L., Ed.; Springer International Publishing: New York, NY, USA, 2018; pp. 165–192. [Google Scholar]
- Hedrick, P.W.; Hedgecock, D. Sex determination: Genetic models for oysters. J. Hered. 2010, 101, 602–611. [Google Scholar] [CrossRef]
- Teaniniuraitemoana, V.; Huvet, A.; Levy, P.; Klopp, C.; Lhuillier, E.; Gaertner-Mazouni, N.; Gueguen, Y.; Moullac, G.L. Gonad transcriptome analysis of pearl oyster Pinctada margaritifera: Identification of potential sex differentiation and sex determining genes. BMC Genom. 2014, 15, 491. [Google Scholar] [CrossRef]
- Zhou, L.Q.; Wang, X.M.; Wu, B.; Sun, X.J.; Chen, S.Q.; Liu, Z.H.; Yang, A.G.; Zhang, S.N.; Zhao, Q.; Zhang, G.W. Chromosome preparation and karyotypes analysis of both male and female Atrina pectinata. Prog. Fish. Sci. 2018, 39, 66–72. [Google Scholar]
- Thiriot-Quiévreux, C. Advances in chromosomal studies of gastropod molluscs. J. Molluscan Stud. 2003, 69, 187–202. [Google Scholar] [CrossRef]
- Guo, X.M.; Allen, S.K. Sex determination and polyploid gigantism in the dwarf surfclam (Mulinia lateralis Say). Genetics 1994, 138, 1199–1206. [Google Scholar] [CrossRef]
- Cai, Y.; Zhou, Y.C.; Feng, Y.Q.; Xie, Z.Y.; Wang, S.F.; Yuan, W. Review of Chromosome Studies of Mollusks in China. J. Trop. Biol. 2014, 5, 297–306. [Google Scholar]
- Xu, K.F.; Li, Q.; Kong, L.F.; Yu, R.H. A first-generation genetic map of the Japanese scallop Patinopecten yessoensis-based AFLP and microsatellite markers. Aquac. Res. 2008, 40, 35–43. [Google Scholar] [CrossRef]
- Zhou, L.Q.; Liu, Z.H.; Dong, Y.H.; Sun, X.S.; Wu, B.; Yu, T.; Zheng, Y.X.; Yang, A.G.; Zhao, Q.; Zhao, D. Transcriptomics analysis revealing candidate genes and networks for sex differentiation of yesso scallop (Patinopecten yessoensis). BMC Genom. 2019, 20, 671. [Google Scholar] [CrossRef]
- Wang, M.; Xia, J.; Jawad, M.; Wei, W.B.; Gui, L.; Liang, X.; Yang, J.L.; Li, M.Y. Transcriptome sequencing analysis of sex-related genes and miRNAs in the gonads of Mytilus coruscus. Front. Mar. Sci. 2022, 9, 1013857. [Google Scholar] [CrossRef]
- Weng, X.X.; Xu, Y.R.; Dong, X.Y.; Luo, X.; You, W.W.; Ke, C.H.; Cai, M.G. Sex-specific markers developed by next-generation sequencing confirmed a male heterogametic sex determination in small abalone, Haliotis diversicolor. Aquaculture 2022, 555, 738256. [Google Scholar] [CrossRef]
- Yue, C.Y.; Li, Q.; Yu, H.; Liu, S.K.; Kong, L.F. Restriction site associated DNA sequencing (RAD-seq) analysis in Pacific oyster Crassostrea gigas based on observation of individual sex changes. Sci. Rep. 2020, 10, 9873. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Liu, L.J.; Zhang, L.J.; Wei, H.L.; Wu, S.X.; Liu, T.; Shu, Y.; Yang, Y.X.; Yang, Z.J.; Wang, S.; et al. Dynamic transcriptome analysis reveals the gene network of gonadal development from the early history life stages in dwarf surfclam Mulinia lateralis. Biol. Sex Differ. 2022, 13, 69. [Google Scholar] [CrossRef]
- Chen, H.; Xiao, G.Q.; Chai, X.L.; Lin, X.G.; Fang, J.; Teng, S.S. Transcriptome analysis of sex-related genes in the blood clam Tegillarca granosa. PLoS ONE 2017, 12, e0184584. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Peng, W.Z.; Wang, Y.; Huang, Z.K.; Feng, Y.; Han, Z.F.; Luo, X.; You, W.W.; Ke, C.H. Identification and dimorphic expression of sex-related genes in Pacific abalone (Haliotis discus hannai). Aquaculture 2023, 574, 739610. [Google Scholar] [CrossRef]
- Yue, C.Y.; Li, Q.; Yu, H. Gonad transcriptome analysis of the Pacific oyster Crassostrea gigas identifies potential genes regulating the sex determination and differentiation process. Mar. Biotechnol. 2018, 20, 206–219. [Google Scholar] [CrossRef]
- Zhang, N.; Xu, F.; Guo, X.M. Genomic analysis of the Pacific oyster (Crassostrea gigas) reveals possible conservation of vertebrate sex determination in a Mollusc. G3 Genes Genomes Genet. 2014, 4, 2207–2217. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, Y.Y.; Zhou, Z.H.; Lin, C.X.; Wei, J.K.; Qin, Y.P.; Xiang, Z.M.; Ma, H.T.; Zhang, Y.; Zhang, Y.H.; et al. Comparative transcriptome analysis of three gonadal development stages reveals potential genes involved in gametogenesis of the fluted giant clam (Tridacna squamosa). BMC Genom. 2020, 21, 872. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.H.; Lin, Z.H.; Dong, Y.H.; Kong, X.H.; He, L.; Xue, L.Y. Gonad transcriptome analysis of the razor clam (Sinonovacula constricta) revealed potential sex-related genes. Front. Mar. Sci. 2021, 8, 725430. [Google Scholar] [CrossRef]
- Kranz, A.M.; Tollenaere, A.; Norris, B.J.; Degnan, B.M.; Degnan, S.M. Identifying the germline in an equally cleaving mollusc: Vasa and Nanos expression during embryonic and larval development of the vetigastropod Haliotis asinina. J. Exp. Zool. B Mol. Dev. Evol. 2010, 314, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Li, Q.; Yu, H. Expression pattern of Piwi-like gene implies the potential role in germline development in the Pacifc oyster Crossosrea gigas. Aquac. Rep. 2020, 18, 100486. [Google Scholar]
- Liu, L.G.; Liu, T.; Wu, S.X.; Li, Y.J.; Wei, H.L.; Zhang, L.J.; Shu, Y.; Yang, Y.X.; Xing, Q.; Wang, S.; et al. Discovery of Nanos1 and Nanos2/3 as germ cell markers during scallop gonadal development. Mar. Biotechnol. 2022, 24, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Zhang, L.L.; Sun, Y.; Ma, X.L.; Wang, J.; Li, R.J.; Zhang, M.W.; Wang, S.; Hu, X.L.; Bao, Z.M. Transcriptome sequencing and comparative analysis of ovary and testis identifes potential key sex-related genes and pathways in scallop Patinopecten yessoensis. Mar. Biotechnol. 2016, 18, 453–465. [Google Scholar] [CrossRef]
- Amor, M.D.; Norman, M.D.; Roura, A.; Leite, T.S.; Gleadall, I.G.; Reid, A.; Perales-Raya, C.; Lu, C.C.; Silvey, C.J.; Vidal, E.A.G.; et al. Morphological assessment of the Octopus vulgaris species complex evaluated in light of molecular-based phylogenetic inferences. Zool. Scr. 2017, 46, 275–288. [Google Scholar] [CrossRef]
- Coffing, G.C.; Tittes, S.; Small, S.T.; Songco-Casey, G.O.; Piscopo, D.M.; Pungor, J.R.; Miller, A.C.; Niell, C.M.; Kern, A.D. Cephalopod sex determination and its ancient evolutionary origin revealed by chromosome-level assembly of the California two-spot Octopus. bioRxiv 2024. bioRxiv:21.581452. [Google Scholar]
- Xu, D.F.; Liu, Y.S.; Chang, Q.; Chen, S.Q.; Zhao, J.J.; Bian, L.; Ge, J.L.; Liu, C.L. Morphology, growth and development in the early life of Octopus vulgaris. Prog. Fish. Sci. 2019, 40, 145–154. [Google Scholar]
- Dan, S.; Iwasaki, H.; Takasugi, A.; Shibasaki, S.; Yamazaki, H.; Oka, M.; Hamasaki, K. Effects of co-supply ratios of swimming crab Portunus trituberculatus zoeae and Artemia on survival and growth of East Asian common octopus Octopus sinensis paralarvae under an upwelling culture system. Aquac. Res. 2019, 50, 1361–1370. [Google Scholar] [CrossRef]
- Li, F.H.; Bian, L.; Ge, J.L.; Han, F.M.; Liu, Z.H.; Li, X.M.; Liu, Y.S.; Lin, Z.S.; Shi, H.L.; Liu, C.L.; et al. Chromosome-level genome assembly of the East Asian common octopus (Octopus sinensis) using PacBio sequencing and Hi-C technology. Mol. Ecol. Resour. 2020, 20, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.Z.; Huang, J.J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research 2013, 2, 188. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Chan, C.K. Analysis of RNA-seq data using TopHat and Cufflinks. Methods Mol. Biol. 2016, 1374, 339–361. [Google Scholar] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D.L. Analysis of relative gene expression data using real-time quantitative PCR and the 2-DDCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hou, R.; Bao, Z.M.; Wang, S.; Su, H.L.; Li, Y.; Du, H.X.; Hu, J.J.; Wang, S.; Hu, X.L. Transcriptome sequencing and De Novo analysis for yesso scallop (Patinopecten yessoensis) using 454 GS FLX. PLoS ONE 2011, 6, e21560. [Google Scholar] [CrossRef]
- Gong, J.W.; Li, Q.; Yu, H.; Liu, S.; Kong, L.F. First de novo transcriptome assembly of Iwagaki oyster, Crassostrea nippona, and comparative evolutionary analysis of salinity-stress response genes in Crassostrea oysters. Mar. Genom. 2021, 56, 100805. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.P.; Zhang, H.W.; Shi, H.H.; Li, Z.J.; Xue, C.H. Application of multi-omics combined with bioinformatics techniques to assess salinity stress response and tolerance mechanisms of Pacific oyster (Crassostrea gigas) during depuration. Fish Shellfish Immunol. 2023, 137, 108779. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.W.; Cao, Y.F.; Li, Z.X.; Jiao, Y.; Du, X.D.; Zheng, Z. Transcriptome analysis reveals the transition and crosslinking of immune response and biomineralization in shell damage repair in pearl oyster. Aquacult. Rep. 2021, 21, 10085. [Google Scholar] [CrossRef]
- Nef, S.; Vassalli, J.D. Complementary pathways in mammalian female sex determination. J. Biol. 2009, 8, 74. [Google Scholar] [CrossRef]
- Li, Q.; Ishikawa, T.O.; Miyoshi, H.; Oshima, M.; Taketo, M.M. A targeted mutation of Nkd1 impairs mouse spermatogenesis. J. Biol. Chem. 2005, 280, 2831–2839. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.F.; Lee-Chang, J.S.; Harris, K.Y.; Sinha-Hikim, A.P.; Rao, M.K. Role of β-catenin in post-meiotic male germ cell differentiation. PLoS ONE 2011, 6, e28039. [Google Scholar] [CrossRef] [PubMed]
- Tomizuka, K.; Horikoshi, K.; Kitada, R.; Sugawara, Y.; Iba, Y.; Kojima, A.; Yoshitome, A.; Yamawaki, K.; Amagai, M.; Inoue, A.; et al. R-spondin1 plays an essential role in ovarian development through positively regulating Wnt-4 signaling. Hum. Mol. Genet. 2008, 17, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, C.; Saito, D.; Nakamura, S.; Sasaki, T.; Asakawa, S.; Shimizu, N.; Mitani, H.; Furutani-Seiki, M.; Tanaka, M.; Kondoh, H. The hotei mutation of medaka in the anti-Müllerian hormone receptor causes the dysregulation of germ cell and sexual development. Proc. Natl. Acad. Sci. USA 2007, 104, 9691–9696. [Google Scholar] [CrossRef]
- Martyniuk, C.J.; Prucha, M.S.; Doperalski, N.J.; Antczak, ap.; Kroll, K.J.; Falciani, F.; Barber, D.S.; Denslow, N.D. Gene expression networks underlying ovarian development in wild largemouth bass (Micropterus salmoides). PLoS ONE 2013, 8, e59093. [Google Scholar] [CrossRef] [PubMed]
- Borggrefe, T.; Oswald, F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell Mol. Life Sci. 2009, 66, 1631–1646. [Google Scholar] [CrossRef] [PubMed]
- Trombly, D.J.; Woodruff, T.K.; Mayo, K.E. Suppression of Notch signaling in the neonatal mouse ovary decreases primordial follicle formation. Endocrinology 2009, 150, 1014–1024. [Google Scholar] [CrossRef]
- Vanorny, D.A.; Prasasya, R.D.; Chalpe, A.J.; Kilen, S.M.; Mayo, K.E. Notch signaling regulates ovarian follicle formation and coordinates follicular growth. Mol. Endocrinol. 2014, 28, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Fu, X.F.; Wang, L.Q.; Wang, J.J.; Ma, H.G.; Cheng, S.F.; Hou, Z.M.; Ma, J.M.; Quan, G.B.; Shen, W.; et al. Primordial follicle assembly was regulated by Notch signaling pathway in the mice. Mol. Biol. Rep. 2014, 41, 1891–1899. [Google Scholar] [CrossRef]
- Ma, Y.W.; Ye, Y.Y.; Yao, R.H.; Qi, P.Z.; Li, J.J. Transcriptome sequencing analysis of sex-related genes in the gonads of Mytilus unguiculatus. Fishes 2023, 8, 456. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Duan, S.H.; Wang, G.L.; Li, J.L. Integrated mRNA and miRNA expression profile analysis of female and male gonads in Hyriopsis cumingii. Sci. Rep. 2021, 11, 665. [Google Scholar] [CrossRef]
- Wei, P.Y.; He, P.P.; Zhang, X.Z.; Li, W.; Zhang, L.; Guan, J.L.; Chen, X.H.; Lin, Y.; Zhuo, X.F.; Li, Q.Z.; et al. Identification and characterization of microRNAs in the gonads of Crassostrea hongkongensis using high-throughput sequencing. Comp. Biochem. Phys. D 2019, 31, 100606. [Google Scholar] [CrossRef] [PubMed]
- Mustapha, U.F.; Jiang, D.N.; Liang, Z.H.; Gu, H.T.; Yang, W.; Chen, H.P.; Deng, S.P.; Wu, T.L.; Tian, C.X.; Zhu, C.H.; et al. Male-specific Dmrt1 is a candidate sex determination gene in spotted scat (Scatophagus argus). Aquaculture 2018, 495, 351–358. [Google Scholar] [CrossRef]
- Romano, S.; Kaufman, O.H.; Marlow, F.L. Loss of dmrt1 restores zebrafish female fates in the absence of cyp19a1a but not rbpms2a/b. Development 2020, 147, dev190942. [Google Scholar] [CrossRef] [PubMed]
- Naimi, A.; Martinez, A.S.; Specq, M.L.; Mrac, B.; Diss, B.; Mathieu, M.; Sourdaine, P. Identification and expression of a factor of the DM family in the oyster Crassostrea gigas. Comp. Biochem. Phys. A 2009, 152, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.F.; Duan, S.H.; Dong, S.S.; Wu, C.D.; Wang, G.L. Molecular characterization and expression analysis of Dmrt1 gene in Hyriopsis cumingii. Genom. Appl. Biol. 2020, 39, 2033–2041. [Google Scholar]
- Hu, F.Q. Study on the Molecular Characteristics and Function of Dmrt1 in Freshwater Pearl Mussel, Hyriopsis schlegelii. Master’s Thesis, Nanchang University, Nanchang, China, 2016. [Google Scholar]
- Li, R.J.; Zhang, L.L.; Li, W.R.; Zhang, Y.; Li, Y.P.; Zhang, M.W.; Zhao, L.; Hu, X.L.; Wang, S.; Bao, Z.M. FOXL2 and DMRT1L are yin and yang genes for determining timing of sex differentiation in the bivalve Mollusk Patinopecten yessoensis. Front. Physiol. 2018, 9, 1166. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, G.; Kaestner, K.H. SnapShot: Forkhead transcription factors I. Cell 2007, 130, 1160. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, G.; Kaestner, K.H. SnapShot: Forkhead transcription factors II. Cell 2007, 131, 192. [Google Scholar] [CrossRef]
- Liu, X.L.; Zhang, Z.F.; Shao, M.Y.; Liu, J.G.; Muhammad, F. Sexually dimorphic expression of foxl2 during gametogenesis in scallop Chlamys farreri, conserved with vertebrates. Dev. Genes Evol. 2012, 222, 279–286. [Google Scholar] [CrossRef]
- Cheung, C.T.; Patinote, A.; Guiguen, Y.; Bobe, J. Foxr1 is a novel maternal-effect gene in fish that is required for early embryonic success. PeerJ 2018, 6, e5534. [Google Scholar] [CrossRef]
- Petit, F.G.; Kervarrec, C.; Jamin, S.P.; Smagulova, F.; Hao, C.X.; Becker, E.; Jégou, B.; Chalmel, F.; Primig, M. Combining RNA and protein profiling data with network interactions identifies genes associated with spermatogenesis in mouse and human. Biol. Reprod. 2015, 92, 1–18. [Google Scholar] [CrossRef]
- Kamachi, Y.; Kondoh, H. Sox proteins: Regulators of cell fate specification and differentiation. Development 2013, 140, 4129–4144. [Google Scholar] [CrossRef] [PubMed]
- Kashimada, K.; Koopman, P. SRY: The master switch in mammalian sex determination. Development 2010, 137, 3921–3930. [Google Scholar] [CrossRef] [PubMed]
- Takehana, Y.; Matsuda, M.; Myosho, T.; Suster, M.L.; Kawakami, K.; Shin, I.T.; Kohara, Y.; Kuroki, Y.; Toyoda, A.; Fujiyama, A.; et al. Co-option of Sox3 as the male-determining factor on the Y chromosome in the fish Oryzias dancena. Nat. Commun. 2014, 5, 4157. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Dong, Y.; Liu, W.B.; Ma, X.X.; Shi, R.H.; Chen, H.Q.; Cui, Z.H.; Ao, L.; Zhang, H.D.; Cao, J.; et al. Epigenetic regulation of Sox30 is associated with testis development in mice. PLoS ONE 2014, 9, e97203. [Google Scholar] [CrossRef] [PubMed]
- Ghiselli, F.; Milani, L.; Chang, P.L.; Hedgecock, D.; Davis, J.P.; Nuzhdin, S.V.; Passamonti, M. De Novo assembly of the manila clam Ruditapes philippinarum transcriptome provides new insights into expression bias, mitochondrial doubly uniparental inheritance and sex determination. Mol. Biol. Evol. 2012, 29, 771–786. [Google Scholar] [CrossRef]
- Yao, C.J.; Wan, H.F.; Zhang, Z.P.; Lin, J.M.; Wang, Y.L. Genome-wide identification and expression profile of the Sox gene family in different tissues and during embryogenesis in the Pacific white shrimp (Litopenaeus vannamei). Gene 2020, 763, 144956. [Google Scholar] [CrossRef]
- Ye, R.H.; Ren, H.B. Molecular clonging and gene expression of Foxl2 and Sox14 gene from Hyriopsis cumingii. Oceanol. Limnol. Sin. 2018, 49, 160–167. [Google Scholar]
- Doniach, T.; Hodgkin, J. A sex-determining gene, fem-1, required for both male and hermaphrodite development in Caenorhabditis elegans. Dev. Biol. 1984, 106, 223–235. [Google Scholar] [CrossRef]
- Ventura-Holman, T.; Lu, D.Y.; Si, X.H.; Izevbigie, E.B.; Maher, J.F. The Fem1c genes: Conserved members of the Fem1 gene family in vertebrates. Gene 2003, 314, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Yin, C.; Chang, Y.Q.; Dou, Y.; Hao, Z.L.; Ding, J. Transcriptome analysis of male and female mature gonads of Japanese scallop Patinopecten yessonsis. Genes Genom. 2016, 38, 1041–1052. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, Y.; Lv, X.J.; Li, S.H.; Li, F.H. Sex biased expression of Fem-1 in larval stages suggests its function in early sex differentiation of Litopeneaus vannamei. Reprod. Breed. 2023, 3, 153–160. [Google Scholar] [CrossRef]
- Li, Y.H.; Sosnik, J.; Brassard, L.; Reese, M.; Spiridonov, N.A.; Bates, T.C.; Johnson, G.R.; Anguita, J.; Visconti, P.E.; Salicioni, A.M. Expression and localization of five members of the testis-specific serine kinase (Tssk) family in mouse and human sperm and testis. Mol. Hum. Reprod. 2010, 17, 42–56. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Forward Primer | Reverse Primer |
|---|---|---|
| β-actin | TGATGGCCAAGTTATCACCA | TGATGGCCAAGTTATCACCA |
| EVM0015963 | AACCTGCTCTTTGCTCTGCAT | CAATGCAGCTGGCTACTGGAC |
| EVM0007741 | TCCCCTTCTAATCAGACCGC | AAGTCAGGAAGGAACTGCTAAC |
| EVM0010793 | CCGTGGTTATGGACAGACTTC | CCCGTTCCTCTTCACTCTTAT |
| EVM0025062 | AGCCGAGCGAACTACAGTACCTC | GGCTGACTGACTTGTACCTCTGC |
| EVM0010330 | ACCGTCCAGGACACACTGAGG | GATCCACTGAGGCAGGCACATG |
| EVM0012836 | TCCCACCTTCTCGTCAGTCT | CCGACTTTGGAGGACATCACC |
| EVM0009749 | CCGTCACAGCTTGATCCAGTCG | TGTGCCGCCGCTAGTCCTG |
| EVM0001451 | ACCAGAACAAGCCAGCGACTTC | CTGCACGGATGCTGTCTGGATAG |
| EVM0015987 | TCCAATCTCAACACTTGCGGCTAC | GAGACACCGGCTGAGCACAAC |
| EVM0023113 | CACCCACGTCCACGACACATG | ACCGAGGGAGGAGTGTGATGTC |
| Sample Name | OSIMT1 | OSIMT2 | OSIMT3 | OSIFO1 | OSIFO2 | OSIFO3 |
|---|---|---|---|---|---|---|
| Clean reads | 45,015,696 | 50,841,250 | 44,986,040 | 61,504,798 | 66,830,622 | 51,194,240 |
| Clean bases | 6.72 | 7.59 | 6.71 | 9.19 | 9.97 | 7.65 |
| GC content (%) | 40.39% | 40.53% | 40.15% | 41.30% | 41.29% | 39.17% |
| ≥Q30 (%) | 93.23% | 92.61% | 92.94% | 93.39% | 93.52% | 93.65% |
| GO ID | Terms | Gene ID | Pfam Annotation | Expression Profile |
|---|---|---|---|---|
| GO:0007548 | sex differentiation | EVM0011468 | DM DNA binding domain | + |
| EVM0021683 | Actin | + | ||
| EVM0026626 | Forkhead domain | - | ||
| GO:0045137 | development of primary sexual characteristics | EVM0021683 | Actin | + |
| EVM0026626 | Forkhead domain | - | ||
| GO:0019953 | sexual reproduction | EVM0010793 | Cytochrome P450 | + |
| EVM0015987 | Helix-loop-helix DNA-binding domain | + | ||
| EVM0025062 | SprT-like family | + | ||
| EVM0001451 | Nucleoside diphosphate kinase; Dpy-30 motif | - | ||
| EVM0007741 | Ca binding region; EF-hand domain; Ca2+ insensitive EF hand | - | ||
| EVM0015963 | 14-3-3 protein | - | ||
| GO:0018992 | germ-line sex determination | EVM0026626 | Forkhead domain | - |
| GO:0046546 | development of primary male sexual characteristics | EVM0026626 | Forkhead domain | - |
| GO:0030238 | male sex determination | EVM0026626 | Forkhead domain | - |
| GO:0007542 | primary sex determination, germ-line | EVM0026626 | Forkhead domain | - |
| GO:0007530 | sex determination | EVM0026626 | Forkhead domain | - |
| GO:0007538 | primary sex determination | EVM0026626 | Forkhead domain | - |
| GO:0019100 | male germ-line sex determination | EVM0026626 | Forkhead domain | - |
| GO:0046661 | male sex differentiation | EVM0026626 | Forkhead domain | - |
| Gene Name | Gene ID | Average FPKM | Functional Annotation | FDR | |
|---|---|---|---|---|---|
| Male | Female | ||||
| Dmrt1 | EVM0011468 | 136.43 | 1.56 | doublesex- and mab-3-related transcription factor 1 | E 3.48 × 10−5 |
| FoxN5 | EVM0026626 | 97.31 | 11.58 | forkhead box protein N-5-like | 1.14 × 10−15 |
| Foxj1 | EVM0024813 | 65.67 | 3.77 | forkhead box protein J1-B-like | 2.18 × 10−15 |
| Sox14 | EVM0023113 | 0.01 | 142.93 | transcription factor Sox-14-like | 2.20 × 10−47 |
| Sox30 | EVM0007650 | 66.79 | 0.40 | transcription factor Sox-30-like | 2.73 × 10−23 |
| Fem1 | EVM0010330 | 18.17 | 3.29 | Sex-determining protein fem-1 | 5.17 × 10−7 |
| Pgk2 | EVM0010209 | 9063.40 | 195.61 | phosphoglycerate kinase 2-like | 1.00 × 10−3 |
| Pde | EVM0008366 | 13.93 | 0 | cGMP-specific 3′,5′-cyclic phosphodiesterase-like | 8.93 × 10−10 |
| Tssk6 | EVM0009652 | 89.66 | 0.01 | testis-specific serine/threonine-protein kinase 6-like | 1.44 × 10−21 |
| Tssk1 | EVM0002859 | 5.38 | 0 | testis-specific serine/threonine-protein kinase 1-like | 9.00 × 10−3 |
| Tssk3 | EVM0024515 | 395.24 | 0.02 | testis-specific serine/threonine-protein kinase 3-like | 9.30 × 10−18 |
| Wdy | EVM0011883 | 50.65 | 6.23 | WD repeat-containing protein on Y chromosome | 9.21 × 10−7 |
| Lrrc34 | EVM0020929 | 101.76 | 0.16 | leucine-rich repeat-containing protein 34-like | 7.25 × 10−15 |
| Nanos3 | EVM0001816 | 2.81 | 66.13 | protein nanos 3 | 1.18 × 10−9 |
| Hsp70 | EVM0023738 | 12.99 | 0.57 | heat shock protein 70 B2-like | 3.18 × 10−15 |
| β-tubulin | EVM0022450 | 85.13 | 3211.12 | tubulin beta chain-like | 8.62 × 10−8 |
| Pabpc1 | EVM0003110 | 0.36 | 829.37 | polyadenylate-binding protein | 9.46 × 10−46 |
| Col4a5 | EVM0025286 | 0.12 | 39.64 | collagen alpha-5(IV) chain-like | 3.99 × 10−10 |
| Col4a2 | EVM0024483 | 10.92 | 130.40 | collagen alpha-2(IV) chain-like | 4.37 × 10−7 |
| Dnmt1 | newGene_40535 | 0.73 | 8.71 | DNA (cytosine-5)-methyltransferase 1-like | 5.40 × 10−10 |
| Suh | EVM0000797 | 1.24 | 10.22 | recombining binding protein suppressor of hairless-like | 4.73 × 10−13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Chen, S.; Zhang, T.; Pan, L.; Liu, C.; Bian, L. Gonadal Transcriptome Sequencing Analysis Reveals the Candidate Sex-Related Genes and Signaling Pathways in the East Asian Common Octopus, Octopus sinensis. Genes 2024, 15, 682. https://doi.org/10.3390/genes15060682
Li F, Chen S, Zhang T, Pan L, Liu C, Bian L. Gonadal Transcriptome Sequencing Analysis Reveals the Candidate Sex-Related Genes and Signaling Pathways in the East Asian Common Octopus, Octopus sinensis. Genes. 2024; 15(6):682. https://doi.org/10.3390/genes15060682
Chicago/Turabian StyleLi, Fenghui, Siqing Chen, Tao Zhang, Luying Pan, Changlin Liu, and Li Bian. 2024. "Gonadal Transcriptome Sequencing Analysis Reveals the Candidate Sex-Related Genes and Signaling Pathways in the East Asian Common Octopus, Octopus sinensis" Genes 15, no. 6: 682. https://doi.org/10.3390/genes15060682
APA StyleLi, F., Chen, S., Zhang, T., Pan, L., Liu, C., & Bian, L. (2024). Gonadal Transcriptome Sequencing Analysis Reveals the Candidate Sex-Related Genes and Signaling Pathways in the East Asian Common Octopus, Octopus sinensis. Genes, 15(6), 682. https://doi.org/10.3390/genes15060682