
Comparative Analysis of Shapley Values Enhances Transcriptomics Insights across Some Common Uterine Pathologies

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Microarray Expression Data Acquisition, Processing, and Exploratory Analysis
2.1.1. Classical Approaches
2.1.2. Comparative Analysis of Shapley Value (CASh) Approach
2.2. Gene Set Enrichment Analysis and Functional Annotation
3. Results
3.1. Datasets and Samples Analyzed
3.2. Functional Enrichment Analysis of the Differentially Expressed Genes
4. Discussion
5. Conclusions
6. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Devesa-Peiro, A.; Sebastian-Leon, P.; Garcia-Garcia, F.; Arnau, V.; Aleman, A.; Pellicer, A.; Diaz-Gimeno, P. Uterine disorders affecting female fertility: What are the molecular functions altered in endometrium? Fertil. Steril. 2020, 113, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Andres, M.P.; Arcoverde, F.V.L.; Souza, C.C.C.; Fernandes, L.F.C.M.; Abrão, M.S.; Kho, R.M. Extrapelvic Endometriosis: A Systematic Review. J. Minim. Invasive Gynecol. 2020, 27, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.S.; Kotlyar, A.M.; Flores, V.A. Endometriosis is a chronic systemic disease: Clinical challenges and novel innovations. Lancet 2021, 397, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Giudice, L.C.; Horne, A.W.; Missmer, S.A. Time for global health policy and research leaders to prioritize endometriosis. Nat. Commun. 2023, 14, 8028. [Google Scholar] [CrossRef] [PubMed]
- Czyzyk, A.; Podfigurna, A.; Szeliga, A.; Meczekalski, B. Update on endometriosis pathogenesis. Minerva Ginecol. 2017, 69, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Crump, J.; Suker, A.; White, L. Endometriosis: A review of recent evidence and guidelines. Aust. J. Gen. Pract. 2024, 53, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, E.; As-Sanie, S.; Marsh, E.E. Epidemiology and management of uterine fibroids. Int. J. Gynaecol. Obstet. 2020, 149, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Somigliana, E.; Vercellini, P.; Daguati, R.; Pasin, R.; De Giorgi, O.; Crosignani, P.G. Fibroids and female reproduction: A critical analysis of the evidence. Hum. Reprod. Update 2007, 13, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.H.; McNally, R.; Kim, J.J.; Wei, J.J. Racial disparity in uterine leiomyoma: New insights of genetic and environmental burden in myometrial cells. Mol. Hum. Reprod. 2024, 30, gaae004. [Google Scholar] [CrossRef]
- Langton, C.R.; Harmon, Q.E.; Baird, D.D. Family History and Uterine Fibroid Development in Black and African American Women. JAMA Netw. Open 2024, 7, e244185. [Google Scholar] [CrossRef]
- Crosbie, E.J.; Kitson, S.J.; McAlpine, J.N.; Mukhopadhyay, A.; Powell, M.E.; Singh, N. Endometrial cancer. Lancet 2022, 399, 1412–1428. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.E.; Bender, D.P. Endometrial Cancer: Obesity, Genetics, and Targeted Agents. Obstet. Gynecol. Clin. N. Am. 2019, 46, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.; Fang, S.; Teichman, P.G.; Xing, L.; Huang, H. Endometrial carcinoma tumorigenesis and pharmacotherapy research. Minerva Endocrinol. 2012, 37, 117–132. [Google Scholar] [PubMed]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E. Endometrial cancer. Lancet 2016, 387, 1094–1108. [Google Scholar] [CrossRef] [PubMed]
- Geng, R.; Huang, X.; Li, L.; Guo, X.; Wang, Q.; Zheng, Y.; Guo, X. Gene expression analysis in endometriosis: Immunopathology insights, transcription factors and therapeutic targets. Front. Immunol. 2022, 13, 1037504. [Google Scholar] [CrossRef] [PubMed]
- Giudice, L.C.; Oskotsky, T.T.; Falako, S.; Opoku-Anane, J.; Sirota, M. Endometriosis in the era of precision medicine and impact on sexual and reproductive health across the lifespan and in diverse populations. FASEB J. 2023, 37, e23130. [Google Scholar] [CrossRef] [PubMed]
- Buyukcelebi, K.; Duval, A.J.; Abdula, F.; Elkafas, H.; Seker-Polat, F.; Adli, M. Integrating leiomyoma genetics, epigenomics, and single-cell transcriptomics reveals causal genetic variants, genes, and cell types. Nat. Commun. 2024, 15, 1169. [Google Scholar] [CrossRef]
- Hever, A.; Roth, R.B.; Hevezi, P.A.; Lee, J.; Willhite, D.; White, E.C.; Marin, E.M.; Herrera, R.; Acosta, H.M.; Acosta, A.J.; et al. Molecular characterization of human adenomyosis. Mol. Hum. Reprod. 2006, 12, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, G.L.; Chandramouli, G.V.; Dainty, L.; Litzi, T.J.; Berchuck, A.; Barrett, J.C.; Risinger, J.I. Microarray analysis of endometrial carcinomas and mixed mullerian tumors reveals distinct gene expression profiles associated with different histologic types of uterine cancer. Clin. Cancer Res. 2005, 11, 4056–4066. [Google Scholar] [CrossRef]
- Risinger, J.I.; Maxwell, G.L.; Chandramouli, G.V.; Jazaeri, A.; Aprelikova, O.; Patterson, T.; Berchuck, A.; Barrett, J.C. Microarray analysis reveals distinct gene expression profiles among different histologic types of endometrial cancer. Cancer Res. 2003, 63, 6–11. [Google Scholar]
- Wang, H.; Mahadevappa, M.; Yamamoto, K.; Wen, Y.; Chen, B.; Warrington, J.A.; Polan, M.L. Distinctive proliferative phase differences in gene expression in human myometrium and leiomyomata. Fertil. Steril. 2003, 80, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Y.; Xiao, Y.; Ruan, J.; Tian, Q.; Cheng, Q.; Chang, K.; Yi, X. Distinct subtypes of endometriosis identified based on stromal-immune microenvironment and gene expression: Implications for hormone therapy. Front. Immunol. 2023, 14, 1133672. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, Q.; Bai, C.; He, K.; Pan, Y. A cross-study gene set enrichment analysis identifies critical pathways in endometriosis. Reprod. Biol. Endocrinol. 2009, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Bryant, P.A.; Venter, D.; Robins-Browne, R.; Curtis, N. Chips with everything: DNA microarrays in infectious diseases. Lancet Infect. Dis. 2004, 4, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Copland, J.A.; Davies, P.J.; Shipley, G.L.; Wood, C.G.; Luxon, B.A.; Urban, R.J. The use of DNA microarrays to assess clinical samples: The transition from bedside to bench to bedside. Recent Prog. Horm. Res. 2003, 58, 25–53. [Google Scholar] [CrossRef] [PubMed]
- Krokidis, M.G.; Vlamos, P. Transcriptomics in amyotrophic lateral sclerosis. Front. Biosci. (Elite Ed.) 2018, 10, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Rai, G.; Rai, R.; Saeidian, A.H.; Rai, M. Microarray to deep sequencing: Transcriptome and miRNA profiling to elucidate molecular pathways in systemic lupus erythematosus. Immunol. Res. 2016, 64, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Ward, K. Microarray technology in obstetrics and gynecology: A guide for clinicians. Am. J. Obstet. Gynecol. 2006, 195, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Esteban, F.J.; Wall, D.P. Using game theory to detect genes involved in Autism Spectrum Disorder. Top 2011, 19, 121–129. [Google Scholar] [CrossRef]
- Jeffery, I.B.; Higgins, D.G.; Culhane, A.C. Comparison and evaluation of methods for generating differentially expressed gene lists from microarray data. BMC Bioinform. 2006, 7, 359. [Google Scholar] [CrossRef]
- Selvaraj, S.; Natarajan, J. Microarray data analysis and mining tools. Bioinformation 2011, 6, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Suhorutshenko, M.; Kukushkina, V.; Velthut-Meikas, A.; Altmäe, S.; Peters, M.; Mägi, R.; Krjutškov, K.; Koel, M.; Codoñer, F.M.; Martinez-Blanch, J.F.; et al. Endometrial receptivity revisited: Endometrial transcriptome adjusted for tissue cellular heterogeneity. Hum. Reprod. 2018, 33, 2074–2086. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Vilella, F.; Alama, P.; Moreno, I.; Mignardi, M.; Isakova, A.; Pan, W.; Simon, C.; Quake, S.R. Single-cell transcriptomic atlas of the human endometrium during the menstrual cycle. Nat. Med. 2020, 26, 1644–1653. [Google Scholar] [CrossRef] [PubMed]
- Breitling, R.; Herzyk, P. Rank-based methods as a non-parametric alternative of the T-statistic for the analysis of biological microarray data. J. Bioinform. Comput. Biol. 2005, 3, 1171–1189. [Google Scholar] [CrossRef] [PubMed]
- Cordero, F.; Botta, M.; Calogero, R.A. Microarray data analysis and mining approaches. Brief. Funct. Genomics 2007, 6, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Patrone, F. Transversality of the Shapley value. TOP 2008, 16, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Sha, G.; Wu, D.; Zhang, L.; Chen, X.; Lei, M.; Sun, H.; Lin, S.; Lang, J. Differentially expressed genes in human endometrial endothelial cells derived from eutopic endometrium of patients with endometriosis compared with those from patients without endometriosis. Hum. Reprod. 2007, 22, 3159–3169. [Google Scholar] [CrossRef] [PubMed]
- Aghajanova, L.; Horcajadas, J.A.; Weeks, J.L.; Esteban, F.J.; Nezhat, C.N.; Conti, M.; Giudice, L.C. The protein kinase A pathway-regulated transcriptome of endometrial stromal fibroblasts reveals compromised differentiation and persistent proliferative potential in endometriosis. Endocrinology 2010, 151, 1341–1355. [Google Scholar] [CrossRef]
- Hodge, J.C.; Park, P.J.; Dreyfuss, J.M.; Assil-Kishawi, I.; Somasundaram, P.; Semere, L.G.; Quade, B.J.; Lynch, A.M.; Stewart, E.A.; Morton, C.C. Identifying the molecular signature of the interstitial deletion 7q subgroup of uterine leiomyomata using a paired analysis. Genes Chromosomes Cancer 2009, 48, 865–885. [Google Scholar] [CrossRef]
- Zavadil, J.; Ye, H.; Liu, Z.; Wu, J.; Lee, P.; Hernando, E.; Soteropoulos, P.; Toruner, G.A.; Wei, J.J. Profiling and functional analyses of microRNAs and their target gene products in human uterine leiomyomas. PLoS ONE 2010, 5, e12362. [Google Scholar] [CrossRef]
- Pappa, K.I.; Polyzos, A.; Jacob-Hirsch, J.; Amariglio, N.; Vlachos, G.D.; Loutradis, D.; Anagnou, N.P. Profiling of Discrete Gynecological Cancers Reveals Novel Transcriptional Modules and Common Features Shared by Other Cancer Types and Embryonic Stem Cells. PLoS ONE 2015, 10, e0142229. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A Comparison of Normalization Methods for High Density Oligonucleotide Array Data Based on Bias and Variance. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Collin, F.; Brettschneider, J.; Simpson, K.; Cope, L.; Irizarry, R.A.; Speed, T.P. Quality Assessment of Affymetrix GeneChip Data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Gentleman, R., Carey, V., Huber, W., Irizarry, R., Dudoit, S., Eds.; Springer: New York, NY, USA, 2005; pp. 33–47. [Google Scholar]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef]
- Pollard, K.S.; Dudoit, S.; van der Laan, M.J. Multiple Testing Procedures: R multtest Package and Applications to Genomics. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Gentleman, R., Carey, V.J., Huber, W., Irizarry, R.A., Dudoit, S., Eds.; Springer: New York, NY, USA, 2005; pp. 249–271. [Google Scholar] [CrossRef]
- Åstrand, M.; Mostad, P.; Rudemo, M. Empirical Bayes models for multiple probe type microarrays at the probe level. BMC Bioinform. 2008, 9, 156. [Google Scholar] [CrossRef]
- Gottardo, R.; Pannucci, J.A.; Kuske, C.R.; Brettin, T. Statistical analysis of microarray data: A Bayesian approach. Biostatistics 2003, 4, 597–620. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Statist. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Moretti, S.; van Leeuwen, D.; Gmuender, H.; Bonassi, S.; van Delft, J.; Kleinjans, J.; Patrone, F.; Merlo, D.F. Combining Shapley value and statistics to the analysis of gene expression data in children exposed to air pollution. BMC Bioinform. 2008, 9, 361. [Google Scholar] [CrossRef]
- Moretti, S. Statistical analysis of the Shapley value for microarray games. Comput. Oper. Res. 2010, 37, 1413–1418. [Google Scholar] [CrossRef]
- Cesari, G.; Algaba, E.; Moretti, S.; Nepomuceno, J.A. An application of the Shapley value to the analysis of co-expression networks. Appl. Netw. Sci. 2018, 3, 35. [Google Scholar] [CrossRef]
- Moretti, S.; Fragnelli, V.; Patrone, F.; Bonassi, S. Using coalitional games on biological networks to measure centrality and power of genes. Bioinformatics 2010, 26, 2721–2730. [Google Scholar] [CrossRef]
- Sun, M.W.; Moretti, S.; Paskov, K.M.; Stockham, N.T.; Varma, M.; Chrisman, B.S.; Washington, P.Y.; Jung, J.Y.; Wall, D.P. Game theoretic centrality: A novel approach to prioritize disease candidate genes by combining biological networks with the Shapley value. BMC Bioinform. 2020, 21, 356. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. g:Profiler—Interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Babu, A.; Ramanathan, G. Multi-omics insights and therapeutic implications in polycystic ovary syndrome: A review. Funct. Integr. Genom. 2023, 23, 130. [Google Scholar] [CrossRef]
- Bonetti, G.; Madeo, G.; Michelini, S.; Ricci, M.; Cestari, M.; Michelini, S.; Gadler, M.; Benedetti, S.; Guerri, G.; Cristofoli, F.; et al. Omics sciences and precision medicine in breast and ovarian cancer. Clin. Ter. 2023, 174, 104–118. [Google Scholar] [CrossRef]
- Boroń, D.; Zmarzły, N.; Wierzbik-Strońska, M.; Rosińczuk, J.; Mieszczański, P.; Grabarek, B.O. Recent Multiomics Approaches in Endometrial Cancer. Int. J. Mol. Sci. 2022, 23, 1237. [Google Scholar] [CrossRef]
- Goulielmos, G.N.; Matalliotakis, M.; Matalliotaki, C.; Eliopoulos, E.; Matalliotakis, I.; Zervou, M.I. Endometriosis research in the -omics era. Gene 2020, 741, 144545. [Google Scholar] [CrossRef]
- Matsuzaki, S. DNA microarray analysis in endometriosis for development of more effective targeted therapies. Front. Biosci. (Elite Ed.) 2011, 3, 1139–1153. [Google Scholar] [CrossRef]
- Shai, R.M. Microarray tools for deciphering complex diseases. Front. Biosci. 2006, 11, 1414–1424. [Google Scholar] [CrossRef]
- Zhao, H.; Jiang, A.; Yu, M.; Bao, H. Identification of biomarkers correlated with diagnosis and prognosis of endometrial cancer using bioinformatics analysis. J. Cell Biochem. 2020, 121, 4908–4921. [Google Scholar] [CrossRef]
- Ajabnoor, G.; Alsubhi, F.; Shinawi, T.; Habhab, W.; Albaqami, W.F.; Alqahtani, H.S.; Nasief, H.; Bondagji, N.; Elango, R.; Shaik, N.A.; et al. Computational approaches for discovering significant microRNAs, microRNA-mRNA regulatory pathways, and therapeutic protein targets in endometrial cancer. Front. Genet. 2023, 13, 1105173. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.J.; Jo, Y.; Cho, M.K.; Jin, J.S.; Kim, J.Y.; Shim, J.; Kim, Y.H.; Park, J.K.; Ryu, D.; Lee, H.J.; et al. Identification and analysis of novel endometriosis biomarkers via integrative bioinformatics. Front. Endocrinol. 2022, 13, 942368. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, S.; Kaneda, K. Genes relating to biological process of endometriosis: Expression changes common to a mouse model and patients. Drug. Res. 2022, 72, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Shen, H.; Ren, X.; Wang, A.; Zhu, S.; Cheng, Y.; Wang, X. Multi-omics analysis reveals the interaction between the complement system and the coagulation cascade in the development of endometriosis. Sci. Rep. 2021, 11, 11926. [Google Scholar] [CrossRef]
- Wang, T.; Jiang, R.; Yao, Y.; Qian, L.; Zhao, Y.; Huang, X. Identification of endometriosis-associated genes and pathways based on bioinformatics analysis. Medicine 2021, 100, e26530. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, L.; Xu, R.; Zhu, C.; Ma, G.; Zhang, C.; Liu, X.; Zhao, H.; Miao, Q. Identification of the molecular relationship between intravenous leiomyomatosis and uterine myoma using RNA sequencing. Sci. Rep. 2019, 9, 1442. [Google Scholar] [CrossRef]

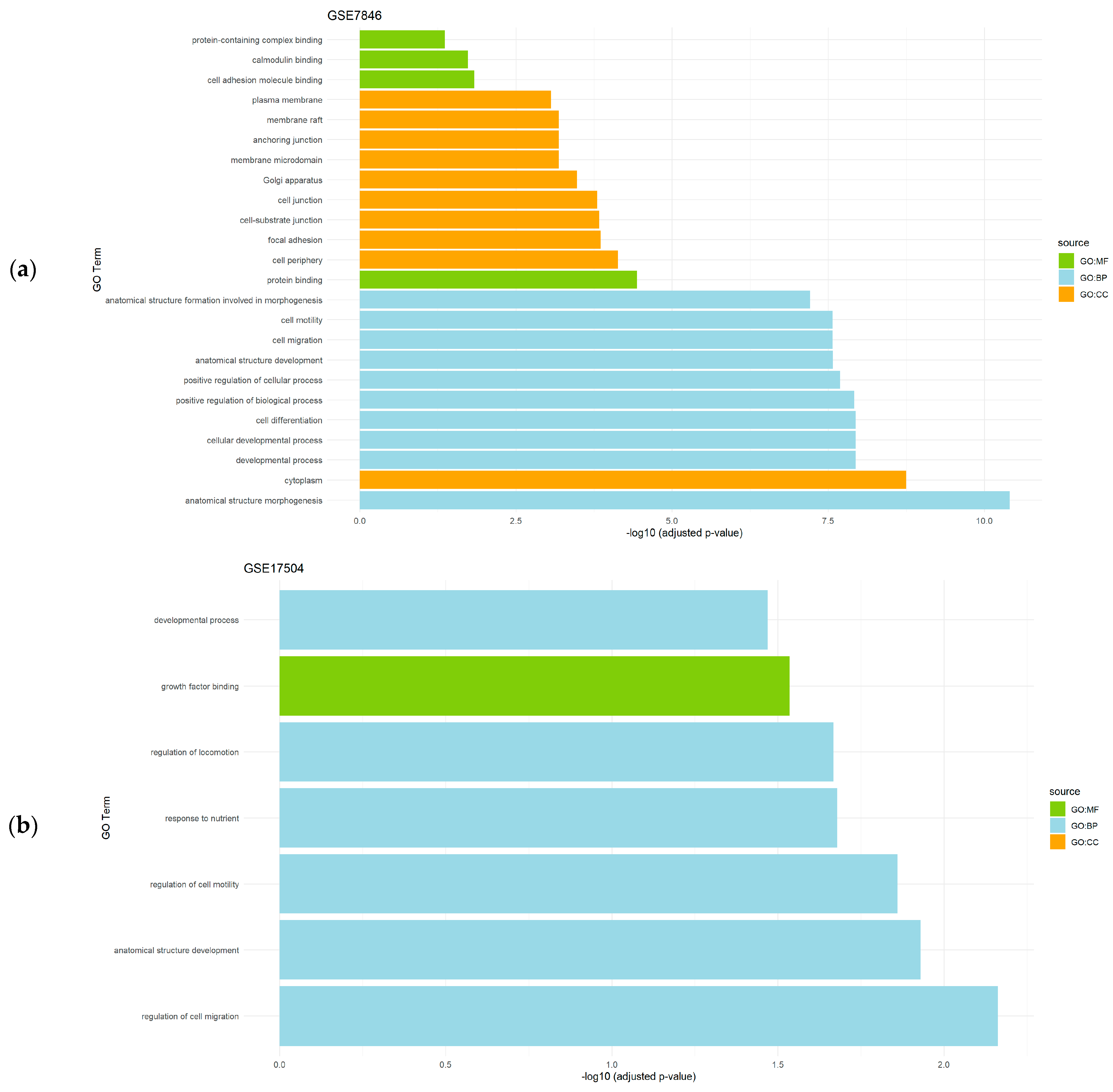


{kind=link}
{kind=link}
{kind=link}
| Phenotype Group | Dataset ID | No. of Samples | Description of Samples |
|---|---|---|---|
| Endometrial cancer | GSE36389 | 16 | Endometrial cancer (n = 10) vs. controls (n = 6) |
| GSE63678 | 11 | Endometrial carcinoma (n = 6) vs. controls (n = 5) | |
| Endometriosis | GSE7846 | 9 | Endometriosis (n = 4) vs. controls (n = 5) |
| GSE17504 | 11 | Endometriosis (n = 5) vs. controls (n = 6) | |
| Uterine leiomyomas | GSE12814 | 14 | Uterine leiomyoma (n = 5) vs. controls (n = 9) |
| GSE23112 | 7 | Uterine leiomyoma (n = 3) vs. controls (n = 4) |
| Dataset ID | Welch’s t-Test | EBayes FDR < 0.01 | EBayes FDR < 0.05 | CASh 0.05 FDR < 0.05 | CASh 0.01 | CASh 0.05 |
|---|---|---|---|---|---|---|
| GSE36389 | 0 | 0 | 0 | 0 | 33 (18 ↑, 15 ↓) | 115 (67 ↑, 48 ↓) |
| GSE63678 | 0 | 0 | 358 | 33 (15 ↑, 18 ↓) | 496 (213 ↑, 283 ↓) | 935 (456 ↑, 479 ↓) |
| GSE7846 | 0 | 0 | 0 | 140 (81↑, 59 ↓) | 71 (39 ↑, 32 ↓) | 333 (194 ↑, 139 ↓) |
| GSE17504 | 0 | 0 | 0 | 17 (12 ↑, 5 ↓) | 17 (9 ↑, 8 ↓) | 83 (49 ↑, 34 ↓) |
| GSE12814 | 0 | 1 | 75 | 0 | 22 (14 ↑, 8 ↓) | 91 (40 ↑, 51 ↓) |
| GSE23112 | 0 | 0 | 0 | 16 (6 ↑, 7 ↓) | 6 (5 ↑, 1 ↓) | 33 (23 ↑, 10 ↓) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro-Martínez, J.A.; Vargas, E.; Díaz-Beltrán, L.; Esteban, F.J. Comparative Analysis of Shapley Values Enhances Transcriptomics Insights across Some Common Uterine Pathologies. Genes 2024, 15, 723. https://doi.org/10.3390/genes15060723
Castro-Martínez JA, Vargas E, Díaz-Beltrán L, Esteban FJ. Comparative Analysis of Shapley Values Enhances Transcriptomics Insights across Some Common Uterine Pathologies. Genes. 2024; 15(6):723. https://doi.org/10.3390/genes15060723
Chicago/Turabian StyleCastro-Martínez, José A., Eva Vargas, Leticia Díaz-Beltrán, and Francisco J. Esteban. 2024. "Comparative Analysis of Shapley Values Enhances Transcriptomics Insights across Some Common Uterine Pathologies" Genes 15, no. 6: 723. https://doi.org/10.3390/genes15060723
APA StyleCastro-Martínez, J. A., Vargas, E., Díaz-Beltrán, L., & Esteban, F. J. (2024). Comparative Analysis of Shapley Values Enhances Transcriptomics Insights across Some Common Uterine Pathologies. Genes, 15(6), 723. https://doi.org/10.3390/genes15060723