
Phylogeny, Genetic Diversity and Population Structure of Fritillaria cirrhosa and Its Relatives Based on Chloroplast Genome Data

Abstract
:1. Introduction
2. Materials and Methods
2.1. Taxon Sampling
2.2. Chloroplast Genome Sequencing, Assembly, and Gene Annotation
2.3. Variation Identification and Statistics
2.4. Phylogenetic and Network Analysis
2.5. Genetic Diversity and Population Differentiation
3. Results
3.1. Feature of the 31 Newly Sequenced Chloroplast Genomes
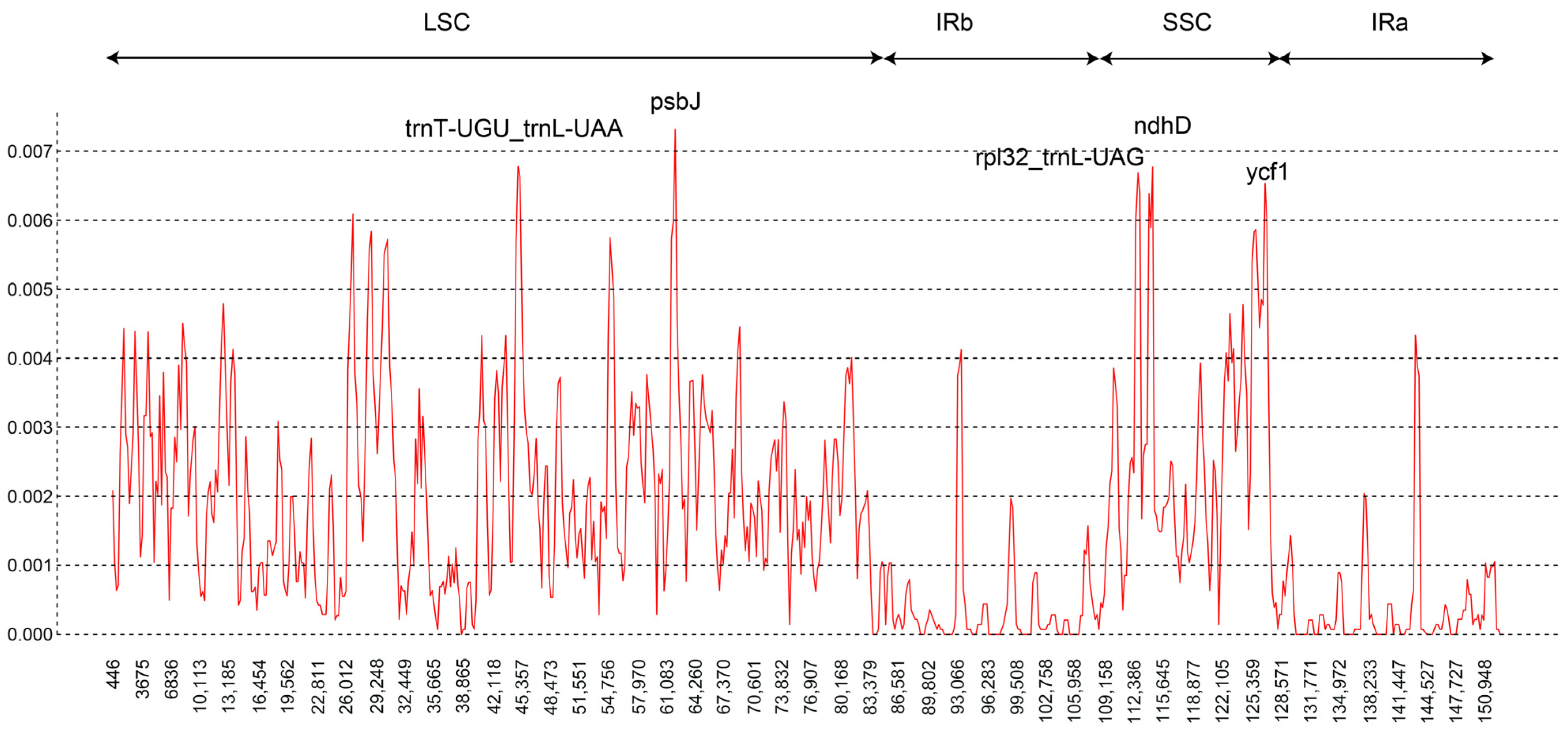
3.2. Chloroplast Genome Sequence Variation
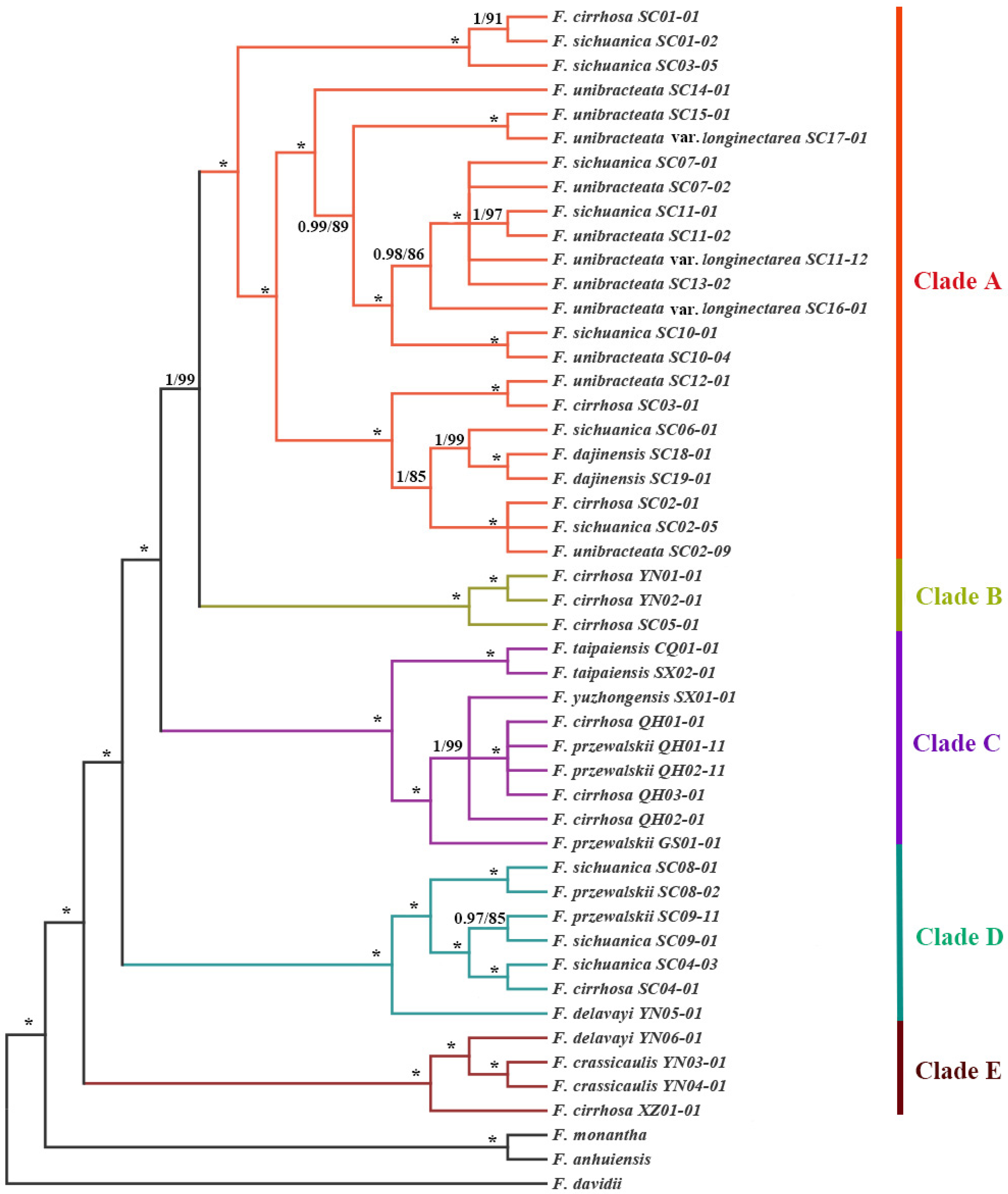
3.3. Phylogenetic Relationships Based on the Chloroplast Genome
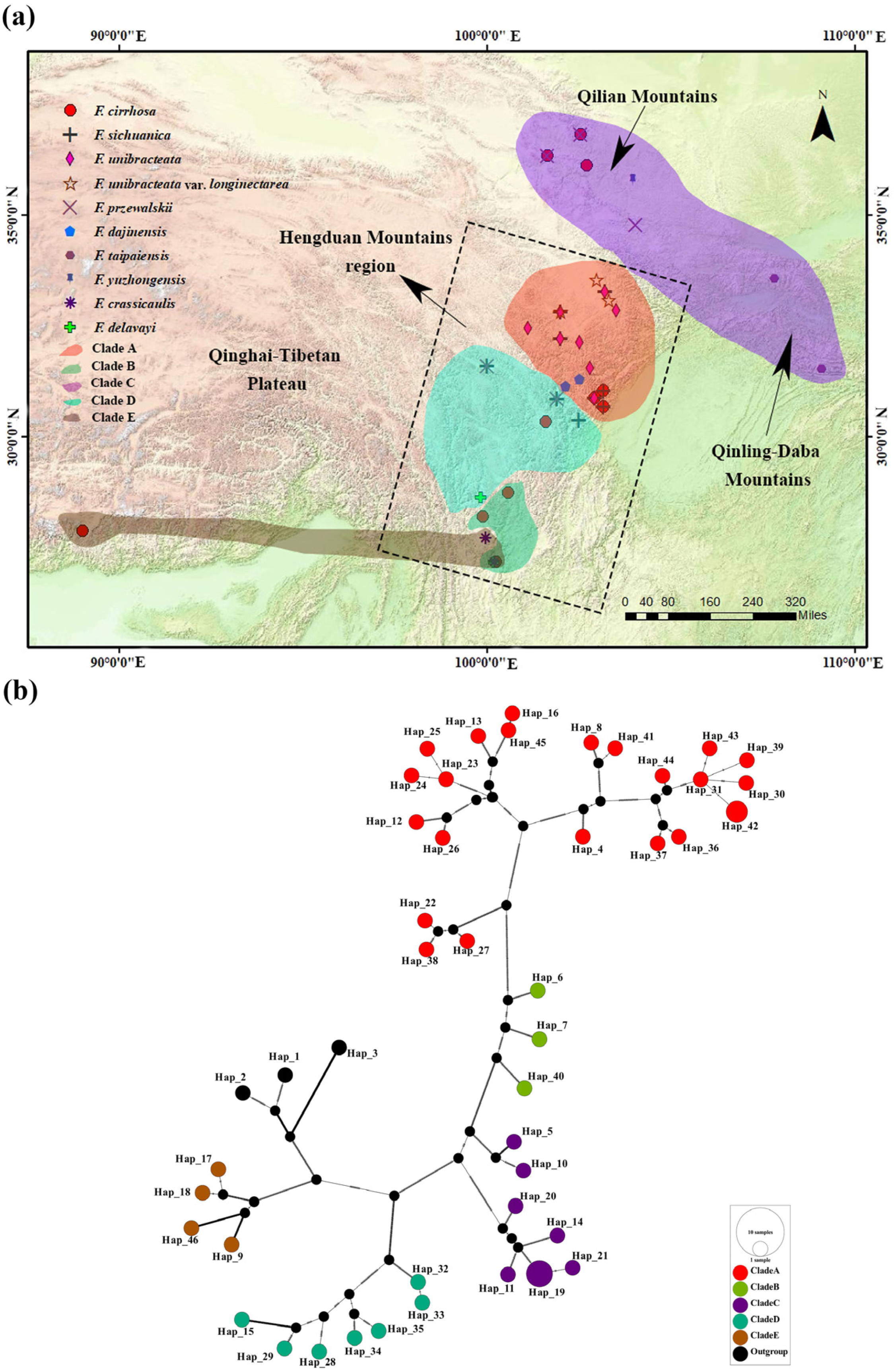
3.4. Population Structure and PCA of F. cirrhosa and Its Relatives
3.5. Genetic Diversity in Different Clades and Species
4. Discussion
4.1. Inter- and Intraspecific Variation in the Chloroplast Genomes of F. cirrhosa and Its Relatives
4.2. Phylogenetic Relationships and Systematic Implications of F. cirrhosa and Its Relatives
4.3. Phylogeographic Structure and Genetic Diversity of F. cirrhosa and Its Relatives
4.4. Conservation Implications for F. cirrhosa and Its Relatives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hao, D.C.; Gu, X.J.; Xiao, P.G.; Peng, Y. Phytochemical and biological research of Fritillaria medicinal resources. Chin. J. Nat. Med. 2013, 11, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Guo, S.H.; Guan, Y.J.; Li, M.; An, Y.; Liu, H. The research progress of medicinal plants Fritillaria. Mol. Plant Breed. 2019, 17, 6198–6206. [Google Scholar]
- Luo, Y.B.; Chen, X.Q. A revision of Fritillaria L. (Liliaceae) in the Hengduan Mountains and adjacent regions, China (1)—A study of Fritillaria cirrhosa D. Don and its related species. Acta Phytotaxon. Sin. 1996, 34, 304–312. [Google Scholar]
- Huang, J.; Yang, L.Q.; Yu, Y.; Liu, Y.M.; Xie, D.F.; Li, J.; He, X.J.; Zhou, S.D. Molecular phylogenetics and historical biogeography of the tribe Lilieae (Liliaceae): Bi-directional dispersal between biodiversity hotspots in Eurasia. Ann. Bot. 2018, 122, 1245–1262. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yu, Y.; Liu, Y.M.; Xie, D.F.; He, X.J.; Zhou, S.D. Comparative chloroplast genomics of Fritillaria (Liliaceae), inferences for phylogenetic relationships between Fritillaria and Lilium and plastome evolution. Plants 2020, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.C.; Che, P.; Zhao, X.L.; Qi, Y.D.; Wei, X.P.; Tang, Z.H.; Zhang, B.G. Resource investigation on Fritillariae cirrhosae bulbus on Tibetan Plateau and its adjacent regions. Mod. Chin. Med. 2021, 23, 611–618, 626. [Google Scholar]
- Xie, J.J.; Tan, P.; Hao, L.; Xiao, Y.; Fang, Q.M.; Zhao, J.N. Development status, strategies and methods of Fritillariae Cirrhosae Bulbus industrial chain based on genetalized science of Chinese material medica. Chin. Tradit. Herb. Drugs 2022, 53, 2150–2163. [Google Scholar]
- Wang, Y.H.; Wang, J.Y.; Garran, T.A.; Liu, H.X.; Lin, H.B.; Luo, J.; Yuan, Q.J.; Sun, J.H.; Dong, W.P.; Guo, L.P. Genetic diversity and population divergence of Leonurus japonicas and its distribution dynamic changes from the last interglacial to the present in China. BMC Plant Biol. 2023, 23, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.B.; Duan, L.Z.; Chen, Q.; Zhang, D.Q. Genetic diversity, population structure, and evolutionary relationships within a taxonomically complex group revealed by AFLP markers: A case study on Fritillaria cirrhosa D. Don and closely related species. Glob. Ecol. Conserv. 2020, 24, e01323. [Google Scholar] [CrossRef]
- Shang, C.; Li, E.Z.; Yu, Z.C.; Lian, M.J.; Chen, Z.; Liu, K.J.; Xu, L.L.; Tong, Z.; Wang, M.F.; Dong, W.P. Chloroplast genomic resources and genetic divergence of endangered species Bretschneidera sinensis (Bretschneideraceae). Front. Ecol. Evol. 2022, 10, 873100. [Google Scholar] [CrossRef]
- Zhang, D.Q.; Gao, L.M.; Yang, Y.P. Genetic diversity and structure of a traditional Chinese medicinal plant species, Fritillaria cirrhosa (Liliaceae) in southwest China and implications for its conservation. Biochem. Systemat. Ecol. 2010, 38, 236–242. [Google Scholar] [CrossRef]
- Li, K.Q.; Wu, W.; Zheng, Y.L.; Dai, Y.; Xiang, L.; Liao, K. Genetic diversity of Fritillaria from Sichuan province based on ISSR. China J. Chin. Mater. Medica 2009, 34, 2149–2154. [Google Scholar]
- Drouin, G.; Daoud, H.; Xia, J. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 2008, 49, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Xu, C.; Wen, J.; Zhou, S. Evolutionary directions of single nucleotide substitutions and structural mutations in the chloroplast genomes of the family Calycanthaceae. BMC Evol. Biol. 2020, 20, 96. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Xu, C.; Wu, P.; Cheng, T.; Yu, J.; Zhou, S.L.; Hong, D.Y. Resolving the systematic positions of enigmatic taxa: Manipulating the chloroplast genome data of Saxifragales. Mol. Phylogenet. Evol. 2018, 126, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Liu, Y.L.; Li, E.Z.; Xu, C.; Sun, J.H.; Li, W.Y.; Zhou, S.L.; Zhang, Z.X.; Suo, Z.L. Phylogenomics and biogeography of Catalpa (Bignoniaceae) reveal incomplete lineage sorting and three dispersal events. Mol. Phylogenet. Evol. 2022, 166, 107330. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.H.; Wang, S.; Wang, Y.H.; Wang, R.S.; Liu, K.J.; Li, E.Z.; Qiao, P.; Shi, L.Y.; Dong, W.P.; Huang, L.Q.; et al. Phylogenomics and genetic diversity of Arnebiae Radix and its allies (Arnebia, Boraginaceae) in China. Front. Plant Sci. 2022, 13, 920826. [Google Scholar] [CrossRef] [PubMed]
- Torre, S.; Sebastiani, F.; Burbui, G.; Pecori, F.; Pepori, A.L.; Passeri, I.; Ghelardini, L.; Selvaggi, A.; Santini, A. Novel Insights Into Refugia at the Southern Margin of the Distribution Range of the Endangered Species Ulmus laevis. Front. Plant Sci. 2022, 13, 826158. [Google Scholar] [CrossRef]
- Xiao, S.Z.; Xu, P.; Deng, Y.T.; Dai, X.B.; Zhao, L.K.; Heider, B.; Zhang, A.; Zhou, Z.L.; Cao, Q.H. Comparative analysis of chloroplast genomes of cultivars and wild species of sweetpotato (Ipomoea batatas [L.] Lam). BMC Genom. 2021, 22, 262. [Google Scholar]
- Huang, D.I.; Hefer, C.A.; Kolosova, N.; Douglas, C.J.; Cronk, Q.C. Whole plastome sequencing reveals deep plastid divergenceand cytonuclear discordance between closely related balsam poplars, Populus balsamifera and P. trichocarpa (Salicaceae). New Phytol. 2014, 204, 693–703. [Google Scholar] [CrossRef]
- Sun, J.H.; Wang, Y.H.; Qiao, P.; Zhao, L.; Li, E.Z.; Dong, W.P.; Zhao, Y.P.; Huang, L.Q. Pueraria Montana population structure and genetic diversity based on chloroplast genome data. Plants 2023, 12, 2231. [Google Scholar] [CrossRef]
- Chen, Q.; Hu, H.S.; Zhang, D.Q. DNA barcoding and phylogenomic analysis of the genus Fritillaria in China based on complete chloroplast genomes. Front. Plant Sci. 2022, 13, 764255. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. Se-Al: Sequence Alignment Editor. Version 2.0 2002, a11. Available online: http://tree.bio.ed.ac.uk/software/seal/ (accessed on 8 August 2002).
- Rozas, J.; Ferrer-Mata, A.; Sanchez-Delbarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mole. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; Bakker, P.I.W.D.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population based linkage analyses. Am. J. Human Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Landis, J.B.; Wang, H.X.; Zhu, Z.X.; Wang, H.F. Comparative analysis of chloroplast genome structure and molecular dating in Myrtales. BMC Plant Biol. 2021, 21, 219. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Xu, C.; Li, C.H.; Sun, J.H.; Zuo, Y.J.; Shi, S.; Cheng, T.; Guo, J.J.; Zhou, S.L. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.S.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Liu, J.; Yu, J.; Wang, L.; Zhou, S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wu, X.B.; Zhang, D.Q. Comparison of the abilities of universal, super, and specific DNA barcodes to discriminate among the original species of Fritillariae cirrhosae bulbus and its adulterants. PLoS ONE 2020, 15, e0229181. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.C.; Wang, Q.; Zhang, P.F.; Araki, H.; Yang, S.H.; Kreitman, M.; Nagylaki, T.; Hudson, R.; Bergelson, J.; Chen, J.Q. Single-nucleotide mutation rate increases close to insertions/deletions in eukaryotes. Nature 2008, 455, U105–U170. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.P.; Toledo, C.A.; Lemmon, E.M.; Lemmon, A.R.; Sytsma, K.J. Out of sight, out of mind: Widespread nuclear and plastid nuclear discordance in the flowering plant genus Polemonium (Polemoniaceae) suggests widespread historical gene flow despite limited nuclear signal. Syst. Biol. 2021, 70, 162–180. [Google Scholar] [CrossRef]
- Stull, G.W.; Pham, K.K.; Soltis, P.S.; Soltis, D.E. Deep reticulation: The long legacy of hybridization in vascular plant evolution. Plant J. 2023, 114, 743–766. [Google Scholar] [CrossRef]
- Wu, J.; Nyman, T.; Wang, D.C.; Argus, G.W.; Yang, Y.P.; Chen, J.H. Phylogeny of Salix subgenus Salix s.l. (Salicaceae): Delimitation, biogeography, and reticulate evolution. BMC Evol. Biol. 2015, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.L.; Li, Z.M.; Mang, X.G.; Sun, W.G. The plastid capture history of the subsect. Campylolepides and the section Ilex (Fagaceae: Quercus). Guihaia 2023, 12, 1–14. [Google Scholar]
- Hill, L. A taxonomic history of Japanese endemic Fritillaria (Liliaceae). Kew. Bull. 2011, 66, 227–240. [Google Scholar] [CrossRef]
- Xie, C.; Xie, D.F.; Zhong, Y.; Guo, X.L.; Liu, Q.; Zhou, S.D.; He, X.J. The effect of Hengduan Mountains Region (HMR) uplift to environmental changes in the HMR and its eastern adjacent area: Tracing the evolutionary history of Allium section Sikkimensia (Amaryllidaceae). Mol. Phylogenetics Evol. 2019, 130, 380–396. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Geng, F.D.; Li, J.J.; Zhang, D.Q.; Gao, F.; Huang, L.; Zhang, X.H.; Kang, J.Q.; Zhang, J.Q.; Ren, Y. Divergence in the Aquilegia ecalcarata complex is correlated with geography and climate oscillations: Evidence from plastid genome data. Mol. Ecol. 2021, 30, 5796–5813. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.A.S.; Bowen, B.W. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. J. Hered. 1998, 89, 415–426. [Google Scholar] [CrossRef]
- Wang, X. Study on the Endangered Mechanism of Angelica sinensis and the Significance of Cultivation on the Protection of Angelica sinensis Resources. Ph.D. Dissertation, Tianjin University of Traditional Chinese Medicine, Tianjing, China, 2020. [Google Scholar]
- Wang, Y.; Sun, J.; Zhao, Z.; Xu, C.; Qiao, P.; Wang, S.; Wang, M.L.; Xu, Z.G.; Yuan, Q.J.; Guo, L.P.; et al. Multiplexed massively parallel sequencing of plastomes provides insights into the genetic diversity, population structure, and phylogeography of wild and cultivated Coptis chinensis. Front. Plant Sci. 2022, 13, 923600. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lee, S.C.; Lee, J.; Lee, H.O.; Joh, H.J.; Kim, N.H.; Park, H.S.; Yang, T.J. Comprehensive Survey of genetic diversity in Chloroplast Genomes and 45S nrDNAs within Panax ginseng species. PLoS ONE 2015, 10, e0117159. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.Q.; Wang, C.M.; Song, B.; Du, F. Corolla retention after pollination facilitates the development of fertilized ovules in Fritillaria delavayi (Liliaceae). Sci. Rep. 2019, 9, 729–738. [Google Scholar] [CrossRef]
- Sajad, B.C.; Behrouz, S.; Masoomeh, K.; Monneni, H.; Hafizi, A.; Khodambashi, M.; Mirakhorli, N.; Sorkheh, K. Assessment of genetic diversity and structure of Imperial Crown (Fritillaria imperialis L.) populations in the Zagros region of Iran using AFLP, ISSR and RAPD markers and implications for its conservation. Biochem. Syst. Ecol. 2012, 42, 35–48. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Polymorphic Sites | Singleton Variable Sites | Parsimony Informative Sites | Indels | Nucleotide Diversity | SNP Density/kb |
|---|---|---|---|---|---|---|
| IR | 71 | 33 | 38 | 20 | 0.00039 | 7.10 |
| LSC | 1178 | 490 | 688 | 363 | 0.00209 | 15.08 |
| SSC | 324 | 122 | 202 | 44 | 0.00288 | 17.12 |
| Total | 1659 | 692 | 967 | 440 | 0.00159 | 13.69 |
| Gene | Protein | Length bp | Nn | Sn | SNP | Density of SNPs/kb |
|---|---|---|---|---|---|---|
| ycf1 | hypothetical protein RF1 | 5523 | 90 | 29 | 119 | 21.55 |
| ndhG | NADH-plastoquinone oxidoreductase subunit 6 | 534 | 66 | 11 | 77 | 144.19 |
| ndhF | NADH-plastoquinone oxidoreductase subunit 5 | 2229 | 19 | 21 | 40 | 17.95 |
| rpoC2 | RNA polymerase β” subunit | 4143 | 15 | 22 | 37 | 8.93 |
| matK | maturase K | 1539 | 16 | 12 | 28 | 18.19 |
| ndhD | NADH-plastoquinone oxidoreductase subunit 4 | 1503 | 8 | 16 | 24 | 15.97 |
| ycf2 | hypothetical protein RF2 | 6654 | 17 | 2 | 19 | 2.86 |
| rps19 | ribosomal protein S19 | 279 | 15 | 4 | 19 | 68.1 |
| atpB | ATP synthase CF1 β subunit | 1497 | 3 | 14 | 17 | 11.36 |
| accD | acetyl-CoA carboxylase carboxyltransferase β subunit | 1464 | 9 | 8 | 17 | 11.61 |
| psbB | photosystem II CP47 chlorophyll apoprotein | 1527 | 3 | 14 | 17 | 11.13 |
| rpoB | RNA polymerase β subunit | 3207 | 8 | 7 | 15 | 4.68 |
| ndhA | NADH-plastoquinone oxidoreductase subunit 1 | 1092 | 6 | 7 | 13 | 11.9 |
| Group | N | S | H | Hd | Pi | Fu’ Fs | Tajima’s D |
|---|---|---|---|---|---|---|---|
| Whole samples | 46 | 1659 | 43 | 0.996 | 0.00159 | −0.22190 | −1.39232 * |
| Clade A | 23 | 486 | 22 | 0.996 | 0.00072 | −0.54245 | −0.62784 |
| Clade B | 3 | 172 | 3 | 1 | 0.00071 | 3.55221 | 0 |
| Clade C | 9 | 281 | 7 | 0.917 | 0.00056 | 5.17739 | −0.67671 |
| Clade D | 7 | 272 | 7 | 1 | 0.00062 | 1.38102 | −0.76924 |
| Clade E | 4 | 385 | 4 | 1 | 0.00134 | 3.49066 | −0.35595 |
| F. cirrhosa | 11 | 980 | 11 | 1 | 0.00168 | 1.41309 | −1.16719 |
| F. sichuanica | 10 | 580 | 10 | 1 | 0.00146 | 1.48401 | 0.41613 |
| F. unibracteata | 8 | 234 | 8 | 1 | 0.00051 | 0.84450 | −0.71048 |
| F. unibracteata var. longinectarea | 3 | 72 | 3 | 1 | 0.00031 | 2.70083 | 0 |
| F. przewalskii | 5 | 410 | 4 | 0.900 | 0.00141 | 7.16923 | 0.97810 |
| F. dajinensis | 2 | 5 | 2 | 1 | 0.00003 | 1.60944 | 0 |
| F. taipaiensis | 2 | 72 | 2 | 1 | 0.00045 | 4.18965 | 0 |
| F. crassicaulis | 2 | 4 | 2 | 1 | 0.00003 | 1.38629 | 0 |
| F. delavayi | 2 | 395 | 2 | 1 | 0.00248 | 5.90536 | 0 |
| Group | Accessions | All Variations | |||
|---|---|---|---|---|---|
| SNPs | Indels | Total | Density/kb | ||
| Clade A | 23 | 486 | 226 | 712 | 4.70 |
| Clade B | 3 | 172 | 60 | 232 | 1.53 |
| Clade C | 9 | 281 | 109 | 390 | 2.57 |
| Clade D | 7 | 272 | 110 | 382 | 2.52 |
| Clade E | 4 | 385 | 115 | 500 | 3.30 |
| Total | 46 | 1659 | 440 | 2099 | 13.85 |
| Comparison | DA | FST (p-Value) |
|---|---|---|
| Clade A vs. Clade B | 0.0016235122 | 0.55553 (0.00000) |
| Clade A vs. Clade C | 0.0017567195 | 0.62007 (0.00000) |
| Clade A vs. Clade D | 0.0021522149 | 0.67751 (0.00000) |
| Clade A vs. Clade E | 0.0022692043 | 0.61952 (0.00000) |
| Clade B vs. Clade C | 0.0017149965 | 0.65073 (0.00000) |
| Clade B vs. Clade D | 0.0020852724 | 0.68970 (0.00000) |
| Clade B vs. Clade E | 0.0022138765 | 0.51357 (0.00592–0.03012) |
| Clade C vs. Clade D | 0.0021363073 | 0.72577 (0.00000) |
| Clade C vs. Clade E | 0.0022374106 | 0.63491 (0.00000) |
| Clade D vs. Clade E | 0.0023211821 | 0.61595 (0.00000) |
| Source of Vatiation | Percentage of Variation | |
|---|---|---|
| AMOVA | Among clades | 63.73 |
| Within clades | 36.27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.; Hu, X.; Zhou, Y.; Peng, Y.-J.; Liu, Z. Phylogeny, Genetic Diversity and Population Structure of Fritillaria cirrhosa and Its Relatives Based on Chloroplast Genome Data. Genes 2024, 15, 730. https://doi.org/10.3390/genes15060730
Huang J, Hu X, Zhou Y, Peng Y-J, Liu Z. Phylogeny, Genetic Diversity and Population Structure of Fritillaria cirrhosa and Its Relatives Based on Chloroplast Genome Data. Genes. 2024; 15(6):730. https://doi.org/10.3390/genes15060730
Chicago/Turabian StyleHuang, Jiao, Xia Hu, Yong Zhou, Yan-Jie Peng, and Zhong Liu. 2024. "Phylogeny, Genetic Diversity and Population Structure of Fritillaria cirrhosa and Its Relatives Based on Chloroplast Genome Data" Genes 15, no. 6: 730. https://doi.org/10.3390/genes15060730