
Rare Pathogenic Variants in Pooled Whole-Exome Sequencing Data Suggest Hyperammonemia as a Possible Cause of Dementia Not Classified as Alzheimer’s Disease or Frontotemporal Dementia

 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Langa, K.M. Cognitive aging, dementia, and the future of an aging population. In Future Directions for the Demography of Aging: Proceedings of a Workshop; National Academies Press (US): Washington, DC, USA, 2018; pp. 249–268. [Google Scholar]
- Kamboh, M.I. Genomics and Functional Genomics of Alzheimer’s Disease. Neurotherapeutics 2022, 19, 152–172. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 66. [Google Scholar] [CrossRef]
- Giau, V.V.; Bagyinszky, E.; Yang, Y.S.; Youn, Y.C.; An, S.S.A.; Kim, S.Y. Genetic analyses of early-onset Alzheimer’s disease using next generation sequencing. Sci. Rep. 2019, 9, 8368. [Google Scholar] [CrossRef]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer’s Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef]
- Chen, H.-H.; Petty, L.E.; Sha, J.; Zhao, Y.; Kuzma, A.; Valladares, O.; Bush, W.; Naj, A.C.; Gamazon, E.R.; Below, J.E.; et al. Genetically regulated expression in late-onset Alzheimer’s disease implicates risk genes within known and novel loci. Transl. Psychiatry 2021, 11, 618. [Google Scholar] [CrossRef]
- König, T.; Stögmann, E. Genetics of Alzheimer’s disease. Wien. Med. Wochenschr. (1946) 2021, 171, 249–256. [Google Scholar] [CrossRef]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef]
- Finger, E.C. Frontotemporal Dementias. Continum 2016, 22, 464–489. [Google Scholar] [CrossRef]
- Rademakers, R.; Neumann, M.; Mackenzie, I.R. Advances in understanding the molecular basis of frontotemporal dementia. Nat. Rev. Neurol. 2012, 8, 423–434. [Google Scholar] [CrossRef]
- Greaves, C.V.; Rohrer, J.D. An update on genetic frontotemporal dementia. J. Neurol. 2019, 266, 2075–2086. [Google Scholar] [CrossRef]
- Olszewska, D.A.; Lonergan, R.; Fallon, E.M.; Lynch, T. Genetics of Frontotemporal Dementia. Curr. Neurol. Neurosci. Rep. 2016, 16, 107. [Google Scholar] [CrossRef]
- Salimi, S.; Irish, M.; Foxe, D.; Hodges, J.R.; Piguet, O.; Burrell, J.R. Visuospatial dysfunction in Alzheimer’s disease and behavioural variant frontotemporal dementia. J. Neurol. Sci. 2019, 402, 74–80. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Neumann, M. Molecular neuropathology of frontotemporal dementia: Insights into disease mechanisms from postmortem studies. J. Neurochem. 2016, 138 (Suppl. S1), 54–70. [Google Scholar] [CrossRef]
- Ferrari, R.; Wang, Y.; Vandrovcova, J.; Guelfi, S.; Witeolar, A.; Karch, C.M.; Schork, A.J.; Fan, C.C.; Brewer, J.B.; International FTD-Genomics Consortium; et al. Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. J. Neurol. Neurosurg. Psychiatry 2017, 88, 152–164. [Google Scholar] [CrossRef]
- Goldman, J.S.; Van Deerlin, V.M. Alzheimer’s Disease and Frontotemporal Dementia: The Current State of Genetics and Genetic Testing Since the Advent of Next-Generation Sequencing. Mol. Diagn. Ther. 2018, 22, 505–513. [Google Scholar] [CrossRef]
- Xiao, X.; Yuan, Z.; Guo, L.; Liao, X.; Zhou, Y.; Zhang, W.; Zhou, L.; Wang, X.; Liu, X.; Liu, H.; et al. The role of frontotemporal dementia associated genes in patients with Alzheimer’s disease. Neurobiol. Aging 2021, 107, 153–158. [Google Scholar] [CrossRef]
- Pasanen, P.; Myllykangas, L.; Pöyhönen, M.; Kiviharju, A.; Siitonen, M.; Hardy, J.; Bras, J.; Paetau, A.; Tienari, P.J.; Guerreiro, R.; et al. Genetics of dementia in a Finnish cohort. Eur. J. Hum. Genet. 2018, 26, 827–837. [Google Scholar] [CrossRef]
- Pagnon de la Vega, M.; Näslund, C.; Brundin, R.; Lannfelt, L.; Löwenmark, M.; Kilander, L.; Ingelsson, M.; Giedraitis, V. Mutation analysis of disease causing genes in patients with early onset or familial forms of Alzheimer’s disease and frontotemporal dementia. BMC Genom. 2022, 23, 99. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Sabbir, M.G.; Albensi, B.C. Ammonia as a Potential Neurotoxic Factor in Alzheimer’s Disease. Front. Mol. Neurosci. 2016, 9, 57. [Google Scholar] [CrossRef]
- Jin, Y.Y.; Singh, P.; Chung, H.J.; Hong, S.T. Blood Ammonia as a Possible Etiological Agent for Alzheimer’s Disease. Nutrients 2018, 10, 564. [Google Scholar] [CrossRef]
- Bobermin, L.D.; Roppa, R.H.A.; Gonçalves, C.-A.; Quincozes-Santos, A. Ammonia-Induced Glial-Inflammaging. Mol. Neurobiol. 2020, 57, 3552–3567. [Google Scholar] [CrossRef]
- Jessy, J.; DeJoseph, M.R.; Hawkins, R.A. Hyperammonaemia depresses glucose consumption throughout the brain. Biochem. J. 1991, 277 Pt 3, 693–696. [Google Scholar] [CrossRef]
- Häberle, J.; Boddaert, N.; Burlina, A.; Chakrapani, A.; Dixon, M.; Huemer, M.; Karall, D.; Martinelli, D.; Crespo, P.S.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J. Rare Dis. 2012, 7, 32. [Google Scholar] [CrossRef]
- Matsumoto, S.; Häberle, J.; Kido, J.; Mitsubuchi, H.; Endo, F.; Nakamura, K. Urea cycle disorders—Update. J. Hum. Genet. 2019, 64, 833–847. [Google Scholar] [CrossRef]
- Rüfenacht, V.; Häberle, J. Mini-Review: Challenges in Newborn Screening for Urea Cycle Disorders. Int. J. Neonatal Screen. 2015, 1, 27–35. [Google Scholar] [CrossRef]
- Rimoin, D.L.; Pyeritz, R.E.; Korf, B.R. Emery and Rimoin’s Principles and Practice of Medical Genetics; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Walker, V. Ammonia toxicity and its prevention in inherited defects of the urea cycle. Diabetes Obes. Metab. 2009, 11, 823–835. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Gorno-Tempini, M.L.; Hillis, A.E.; Weintraub, S.; Kertesz, A.; Mendez, M.; Cappa, S.F.; Ogar, J.M.; Rohrer, J.D.; Black, S.; Boeve, B.F.; et al. Classification of primary progressive aphasia and its variants. Neurology 2011, 76, 1006–1014. [Google Scholar] [CrossRef]
- Rascovsky, K.; Grossman, M. Clinical diagnostic criteria and classification controversies in frontotemporal lobar degeneration. Int. Rev. Psychiatry 2013, 25, 145–158. [Google Scholar] [CrossRef]
- Chang, X.; Wang, K. wANNOVAR: Annotating genetic variants for personal genomes via the web. J. Med. Genet. 2012, 49, 433–436. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2005, 22, 195–201. [Google Scholar] [CrossRef]
- Karlberg, T.; Collins, R.; van den Berg, S.; Flores, A.; Hammarström, M.; Högbom, M.; Holmberg Schiavone, L.; Uppenberg, J. Structure of human argininosuccinate synthetase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 64, 279–286. [Google Scholar] [CrossRef]
- Kimani, J.K.; Wei, T.; Chol, K.; Li, Y.; Yu, P.; Ye, S.; Huang, X.; Qi, M. Functional analysis of novel splicing and missense mutations identified in the ASS1 gene in classical citrullinemia patients. Clin. Chim. Acta 2015, 438, 323–329. [Google Scholar] [CrossRef]
- Sen, K.; Whitehead, M.; Castillo Pinto, C.; Caldovic, L.; Gropman, A. Fifteen years of urea cycle disorders brain research: Looking back, looking forward. Anal. Biochem. 2022, 636, 114343. [Google Scholar] [CrossRef]
- Berning, C.; Bieger, I.; Pauli, S.; Vermeulen, T.; Vogl, T.; Rummel, T.; Höhne, W.; Koch, H.G.; Rolinski, B.; Gempel, K.; et al. Investigation of citrullinemia type I variants by in vitro expression studies. Hum. Mutat. 2008, 29, 1222–1227. [Google Scholar] [CrossRef]
- Kose, E.; Unal, O.; Bulbul, S.; Gunduz, M.; Häberle, J.; Arslan, N. Identification of three novel mutations in fourteen patients with citrullinemia type 1. Clin. Biochem. 2017, 50, 686–689. [Google Scholar] [CrossRef]
- Lisjak, M.; Iaconcig, A.; Guarnaccia, C.; Vicidomini, A.; Moretti, L.; Collaud, F.; Ronzitti, G.; Zentilin, L.; Muro, A.F. Lethality rescue and long-term amelioration of a citrullinemia type I mouse model by neonatal gene-targeting combined to SaCRISPR-Cas9. Mol. Ther.-Methods Clin. Dev. 2023, 31, 101103. [Google Scholar] [CrossRef]
- Daou, M.; Souaid, M.; Yammine, T.; Khneisser, I.; Mansour, H.; Salem, N.; Nemr, A.; Awwad, J.; Moukarzel, A.; Farra, C. Analysis of ASS1 gene in ten unrelated middle eastern families with citrullinemia type 1 identifies rare and novel variants. Mol. Genet. Genom. Med. 2023, 11, e2058. [Google Scholar] [CrossRef]
- Diez-Fernandez, C.; Rüfenacht, V.; Häberle, J. Mutations in the Human Argininosuccinate Synthetase (ASS1) Gene, Impact on Patients, Common Changes, and Structural Considerations. Hum. Mutat. 2017, 38, 471–484. [Google Scholar] [CrossRef]
- Dasarathy, S.; Mookerjee, R.P.; Rackayova, V.; Rangroo Thrane, V.; Vairappan, B.; Ott, P.; Rose, C.F. Ammonia toxicity: From head to toe? Metab. Brain Dis. 2017, 32, 529–538. [Google Scholar] [CrossRef]
- Kalyta, K.; Stelmaszczyk, W.; Szczęśniak, D.; Kotuła, L.; Dobosz, P.; Mroczek, M. The Spectrum of the Heterozygous Effect in Biallelic Mendelian Diseases—The Symptomatic Heterozygote Issue. Genes 2023, 14, 1562. [Google Scholar] [CrossRef]
- Raabe, W. Effects of Hyperammonemia on Neuronal Function: NH4+, IPSP and Cl−-Extrusion. In Cirrhosis, Hyperammonemia, and Hepatic Encephalopathy; Grisolía, S., Felipo, V., Eds.; Springer US: Boston, MA, USA, 1993; pp. 71–82. [Google Scholar]
- Ghosh, S.; Durgvanshi, S.; Agarwal, S.; Raghunath, M.; Sinha, J.K. Current Status of Drug Targets and Emerging Therapeutic Strategies in the Management of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 883–903. [Google Scholar] [CrossRef]
- Lemberg, A.; Alejandra Fernández, M. Hepatic encephalopathy, ammonia, glutamate, glutamine and oxidative stress. Ann. Hepatol. 2009, 8, 95–102. [Google Scholar] [CrossRef]
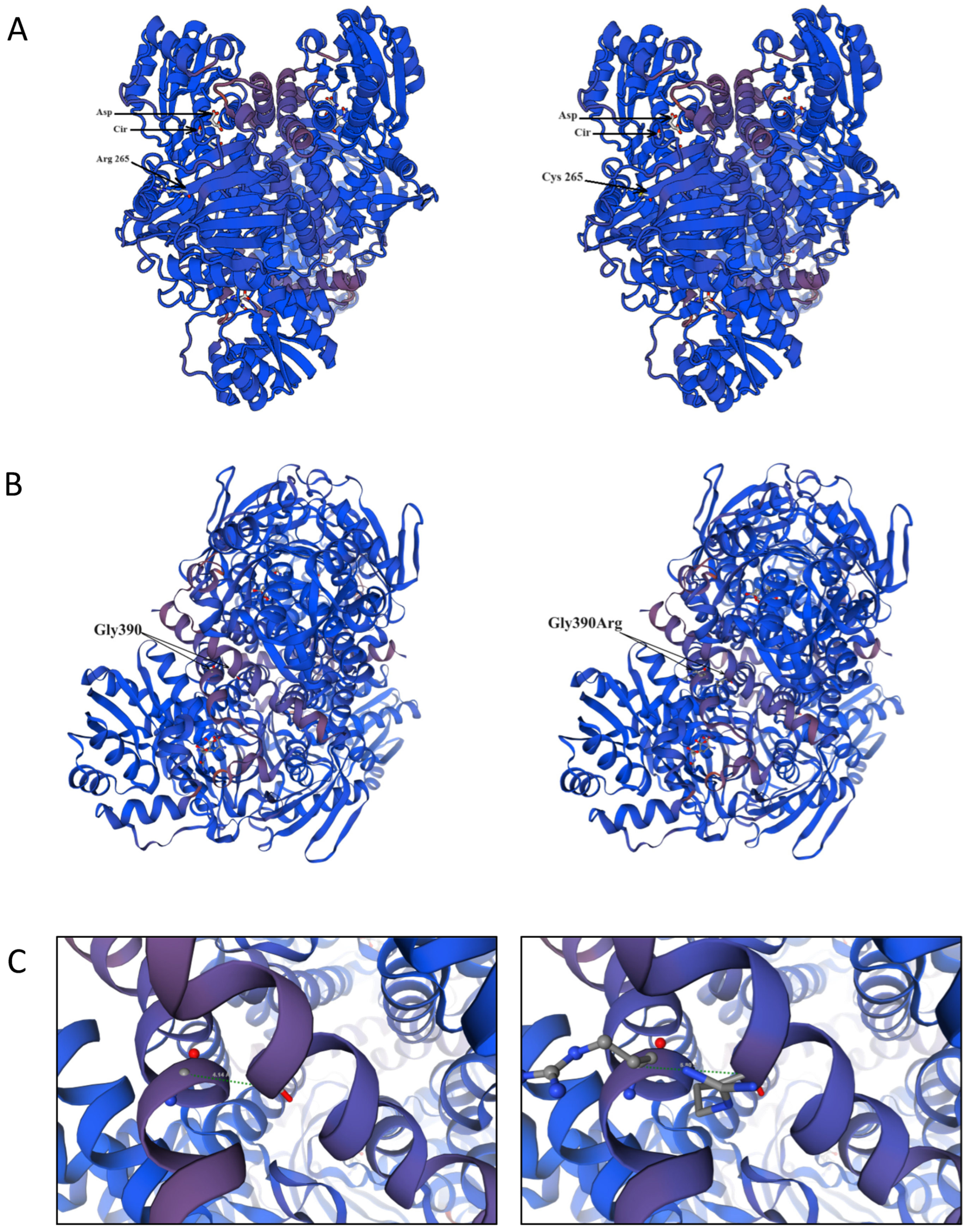
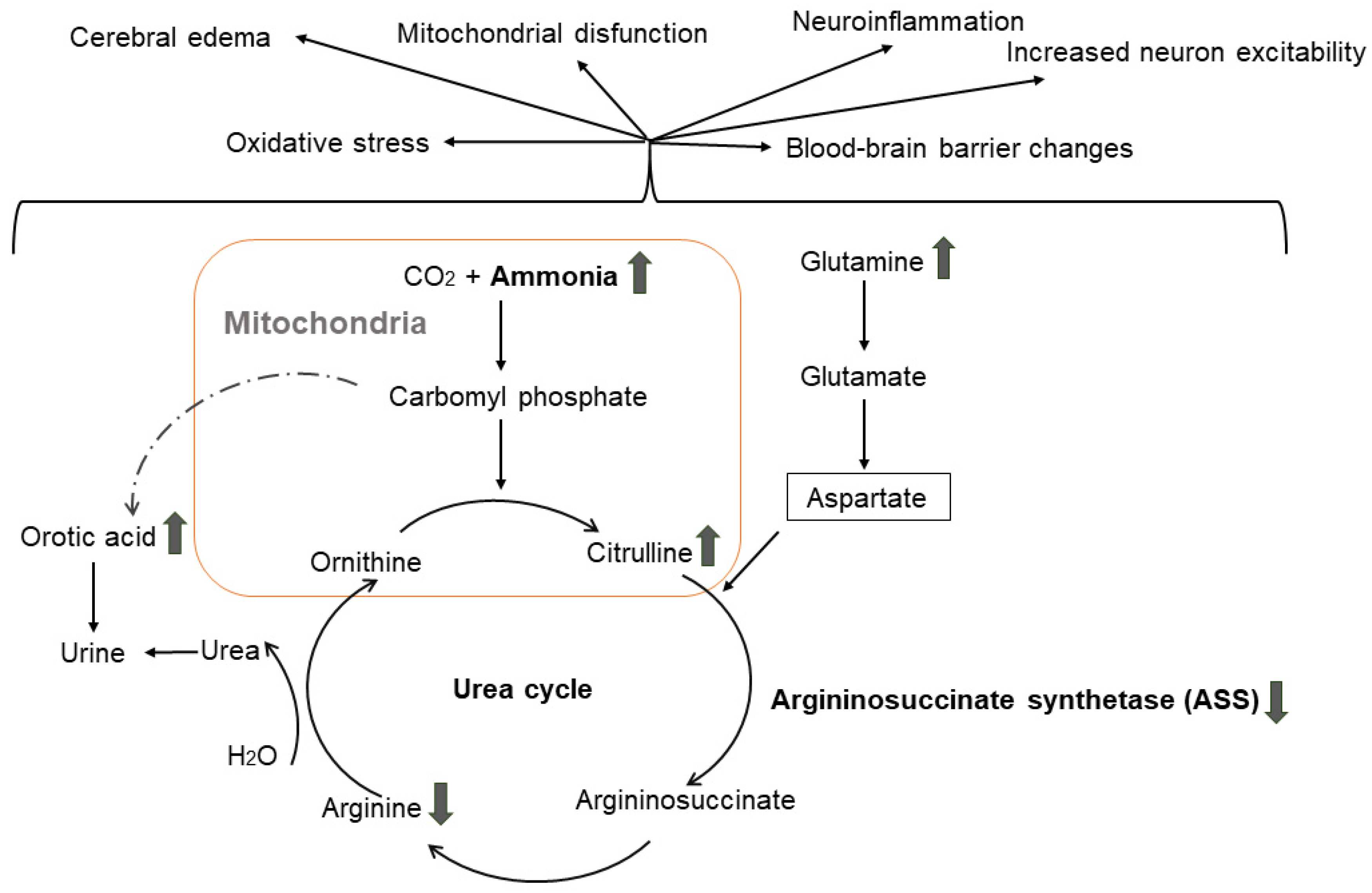

{kind=link}
{kind=link}
| Variant | Current Study | Bulgarian gnomAD Exomes, v.2.1.1 | p-Value | ||||
|---|---|---|---|---|---|---|---|
| Allele Count | Allele Number | MAF (%) | Allele Count | Allele Number | MAF (%) | ||
| rs148918985, ASS1, C>T, p.Arg265Cys | 4 | 807 | 0.50 | 0 | 2666 | 0 | 0.002457 |
| rs121908641, G>A, ASS1, p.Gly390Arg | 13 | 1032 | 1.26 | 2 | 2568 | 0.08 | 0.000002218 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karachanak-Yankova, S.; Serbezov, D.; Antov, G.; Stancheva, M.; Mihaylova, M.; Hadjidekova, S.; Toncheva, D.; Pashov, A.; Belejanska, D.; Zhelev, Y.; et al. Rare Pathogenic Variants in Pooled Whole-Exome Sequencing Data Suggest Hyperammonemia as a Possible Cause of Dementia Not Classified as Alzheimer’s Disease or Frontotemporal Dementia. Genes 2024, 15, 753. https://doi.org/10.3390/genes15060753
Karachanak-Yankova S, Serbezov D, Antov G, Stancheva M, Mihaylova M, Hadjidekova S, Toncheva D, Pashov A, Belejanska D, Zhelev Y, et al. Rare Pathogenic Variants in Pooled Whole-Exome Sequencing Data Suggest Hyperammonemia as a Possible Cause of Dementia Not Classified as Alzheimer’s Disease or Frontotemporal Dementia. Genes. 2024; 15(6):753. https://doi.org/10.3390/genes15060753
Chicago/Turabian StyleKarachanak-Yankova, Sena, Dimitar Serbezov, Georgi Antov, Mikaela Stancheva, Marta Mihaylova, Savina Hadjidekova, Draga Toncheva, Anastas Pashov, Diyana Belejanska, Yavor Zhelev, and et al. 2024. "Rare Pathogenic Variants in Pooled Whole-Exome Sequencing Data Suggest Hyperammonemia as a Possible Cause of Dementia Not Classified as Alzheimer’s Disease or Frontotemporal Dementia" Genes 15, no. 6: 753. https://doi.org/10.3390/genes15060753
APA StyleKarachanak-Yankova, S., Serbezov, D., Antov, G., Stancheva, M., Mihaylova, M., Hadjidekova, S., Toncheva, D., Pashov, A., Belejanska, D., Zhelev, Y., Petrova, M., Mehrabian, S., & Traykov, L. (2024). Rare Pathogenic Variants in Pooled Whole-Exome Sequencing Data Suggest Hyperammonemia as a Possible Cause of Dementia Not Classified as Alzheimer’s Disease or Frontotemporal Dementia. Genes, 15(6), 753. https://doi.org/10.3390/genes15060753