

Integrated Analysis of scRNA-Seq and Bulk RNA-Seq Reveals Metabolic Reprogramming of Liver Cancer and Establishes a Prognostic Risk Model

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
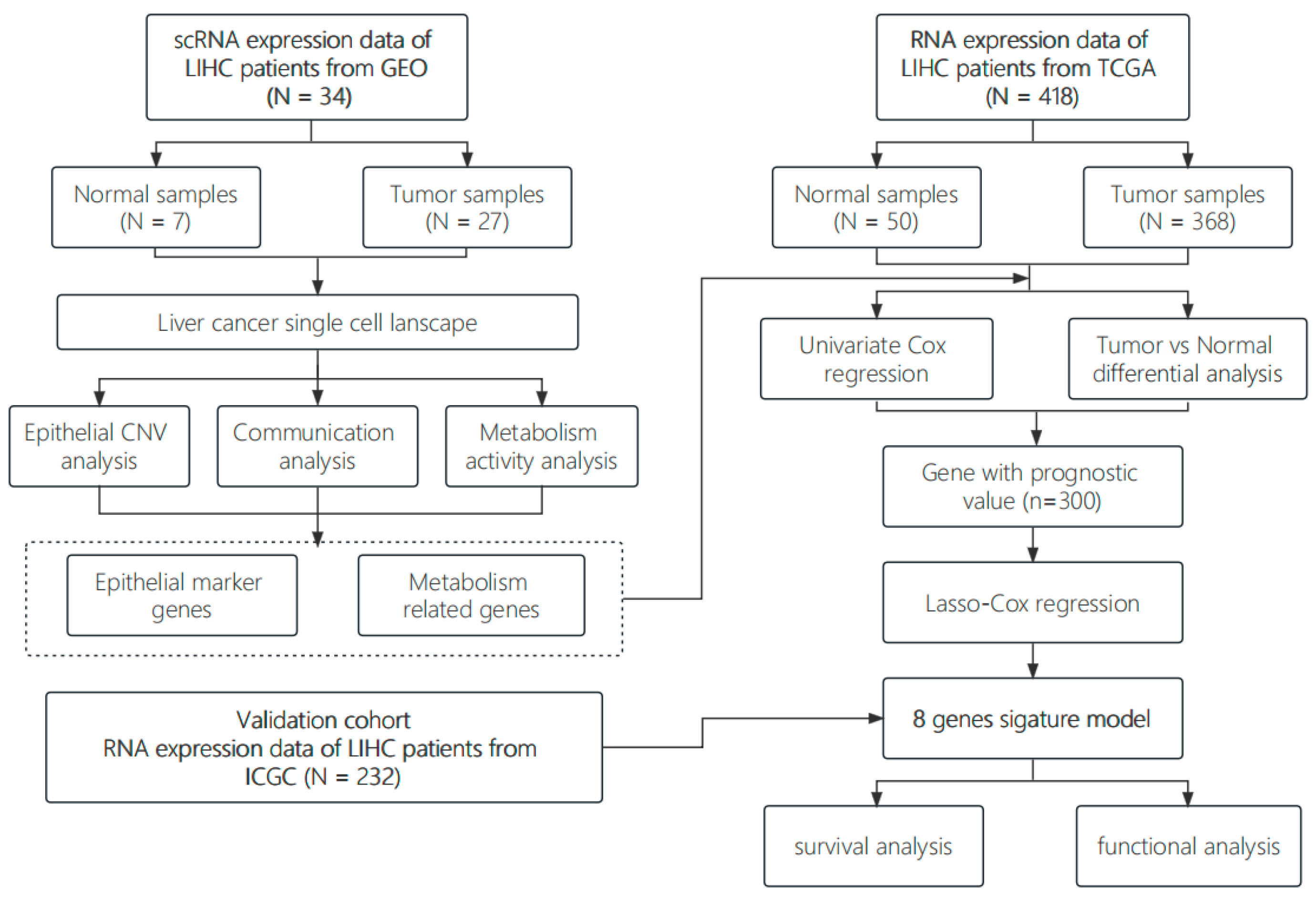
2. Materials and Methods
2.1. Single Cell Data Acquisition and Preprocessing
2.2. Cell Clustering and Annotation
2.3. Copy Number Variation Analysis
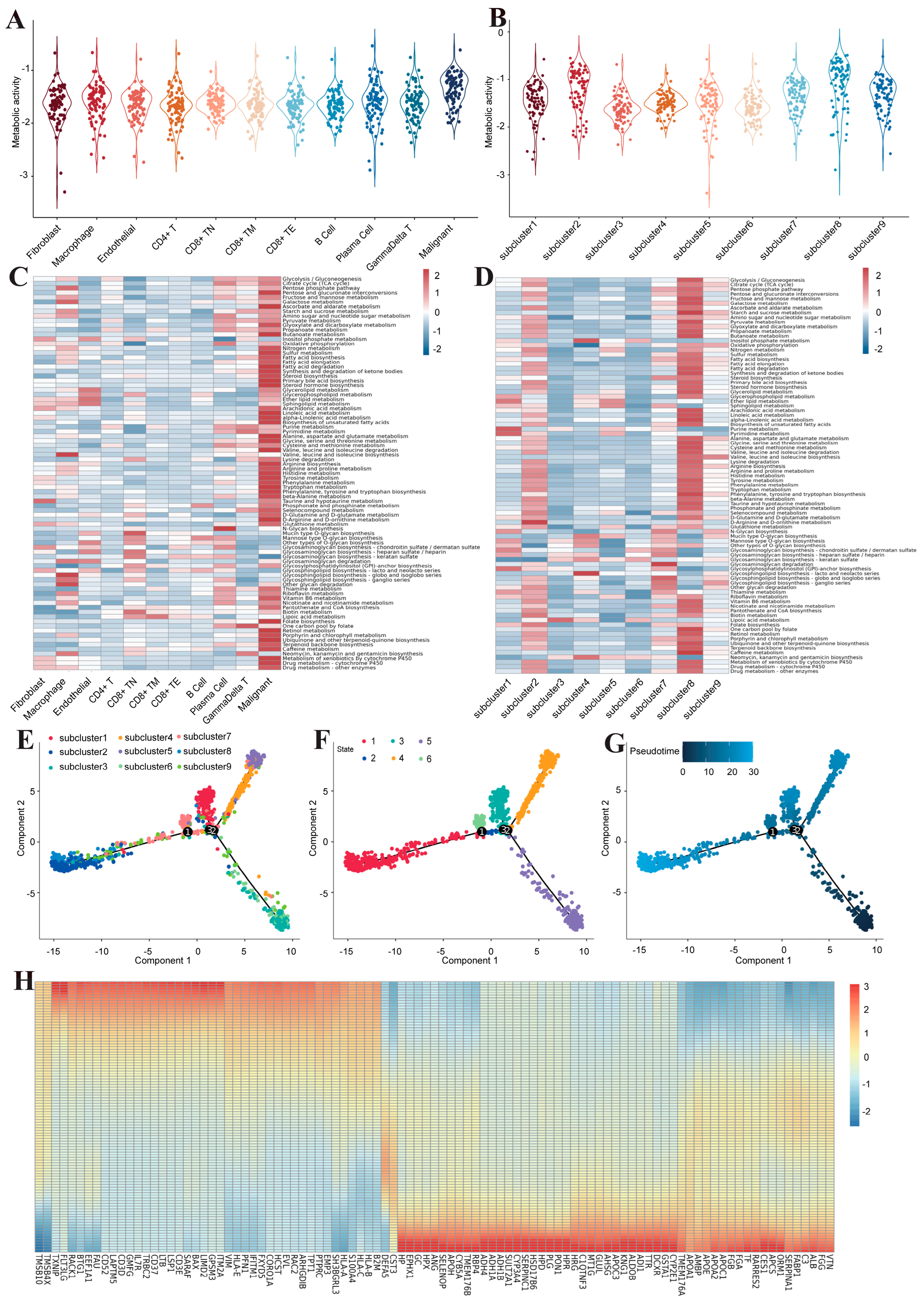
2.4. Gene Set Variation Analysis and Metabolic Activity Analysis
2.5. Cell Communication Analysis
2.6. RNA-Seq Data Acquisition and Preprocessing
2.7. Predictive Model Construction and Validation
2.8. Immune Infiltration Analysis
2.9. Drug Sensitivity Analysis
3. Results
3.1. Construction of a Single-Cell Atlas for Human Liver Cancer
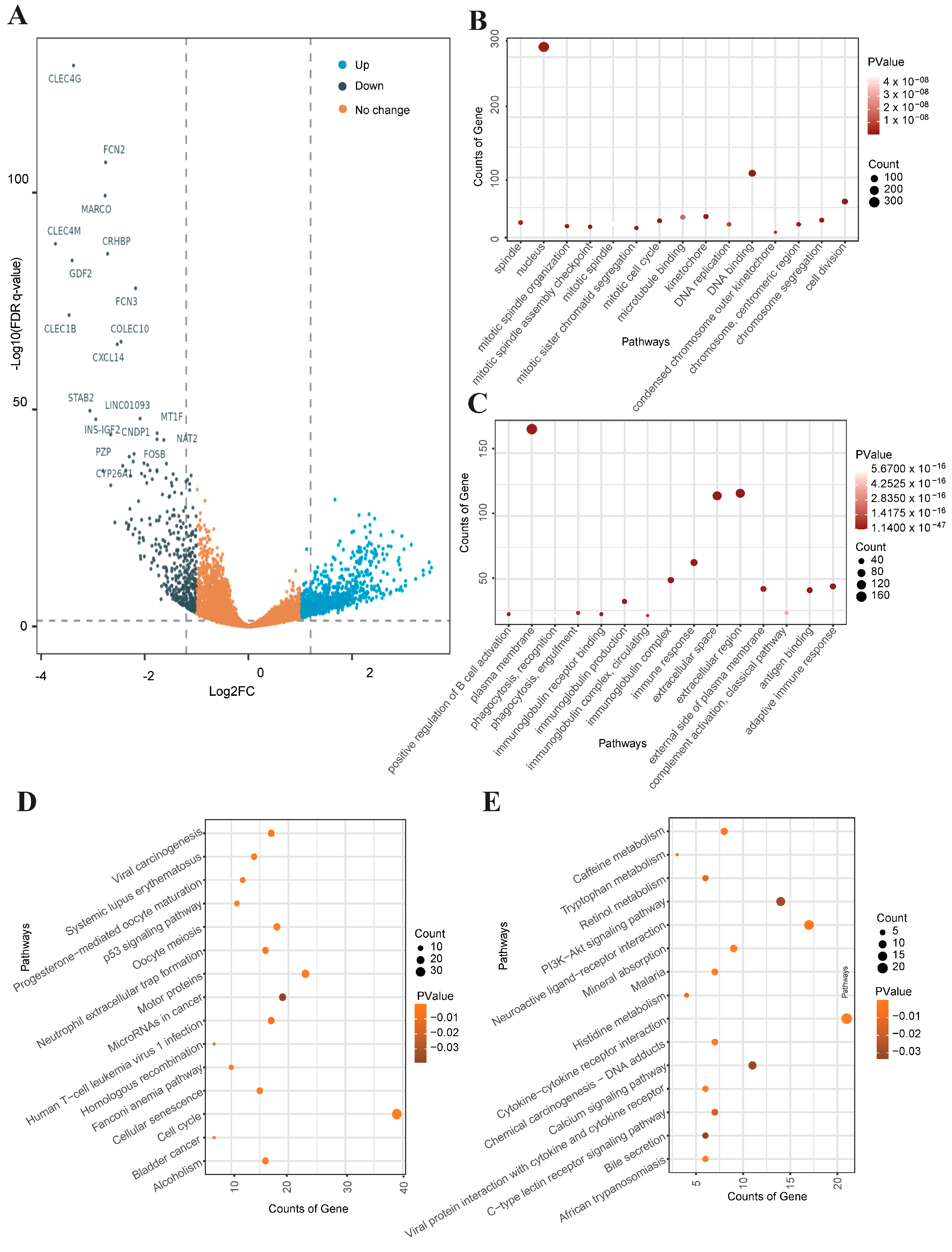
3.2. Inter-Transcriptome Heterogeneity of Human Liver Malignant Cells
3.3. Metabolic Reprogramming of Malignant Cells in Human Liver Cancer
3.4. The MIF/CD74 Axis Is Rich in Interactions between Malignant Cells and Other Cells
3.5. Identification and Functional Enrichment Analysis of DEGs in Liver Cancer RNA-Seq Data
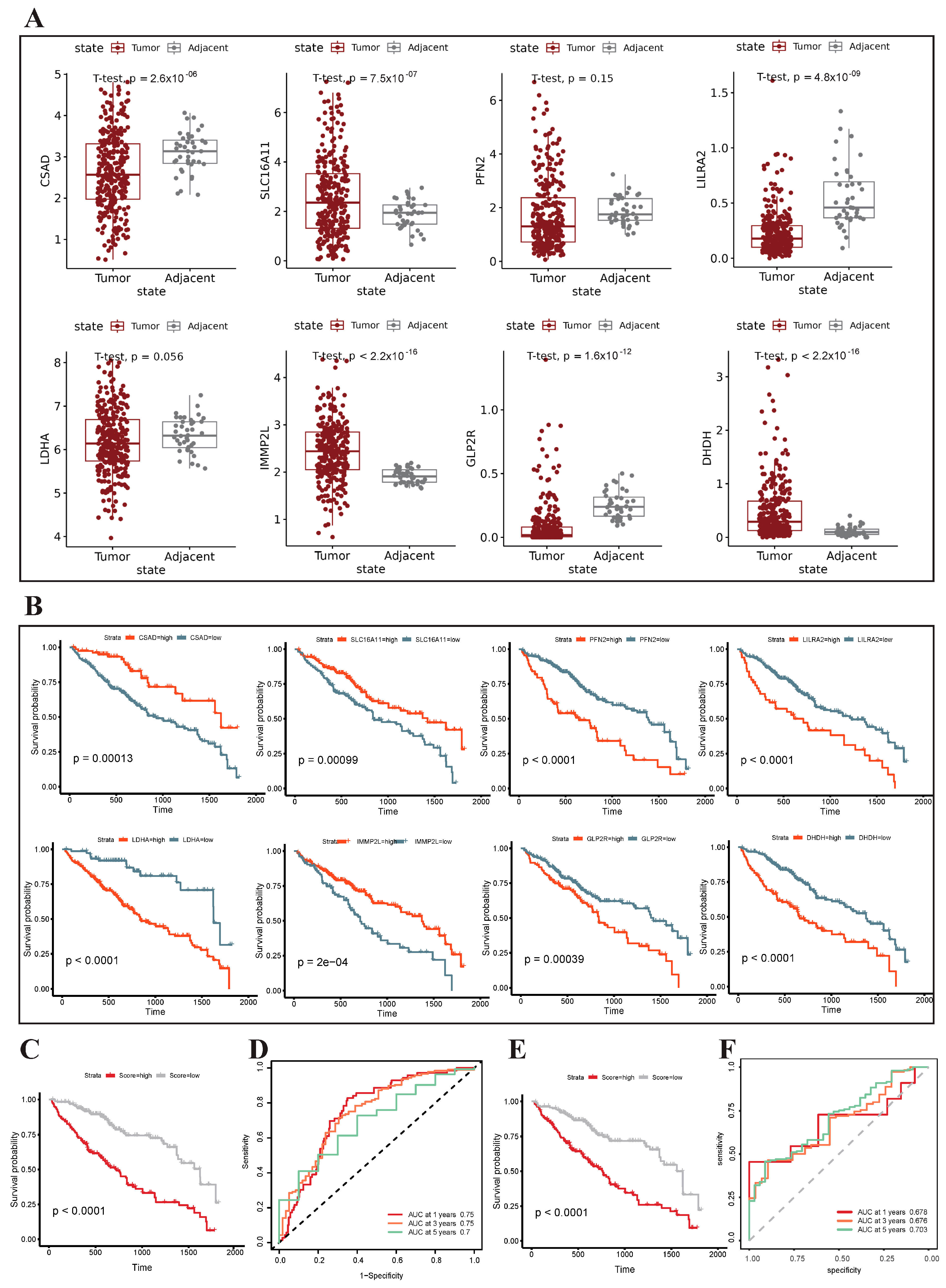
3.6. Construction and Validation of the Metabolism-Related Prognostic Risk Model
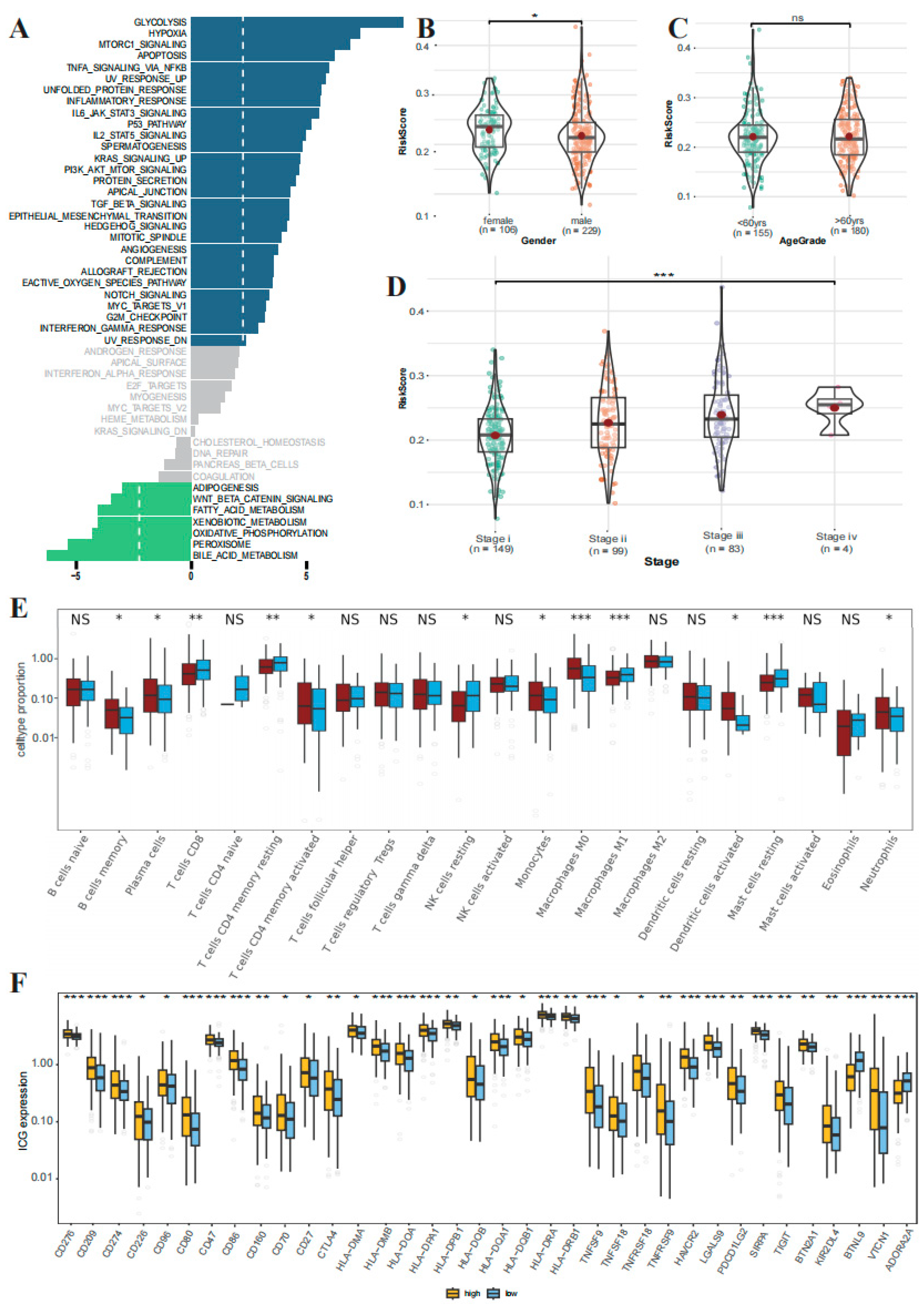
3.7. GSEA Analysis and Clinical Correlation between the High- and Low-Risk Groups
3.8. The Correlations Immune Cell Infiltration and Immune Checkpoint Expression between the High- and Low-Risk Groups
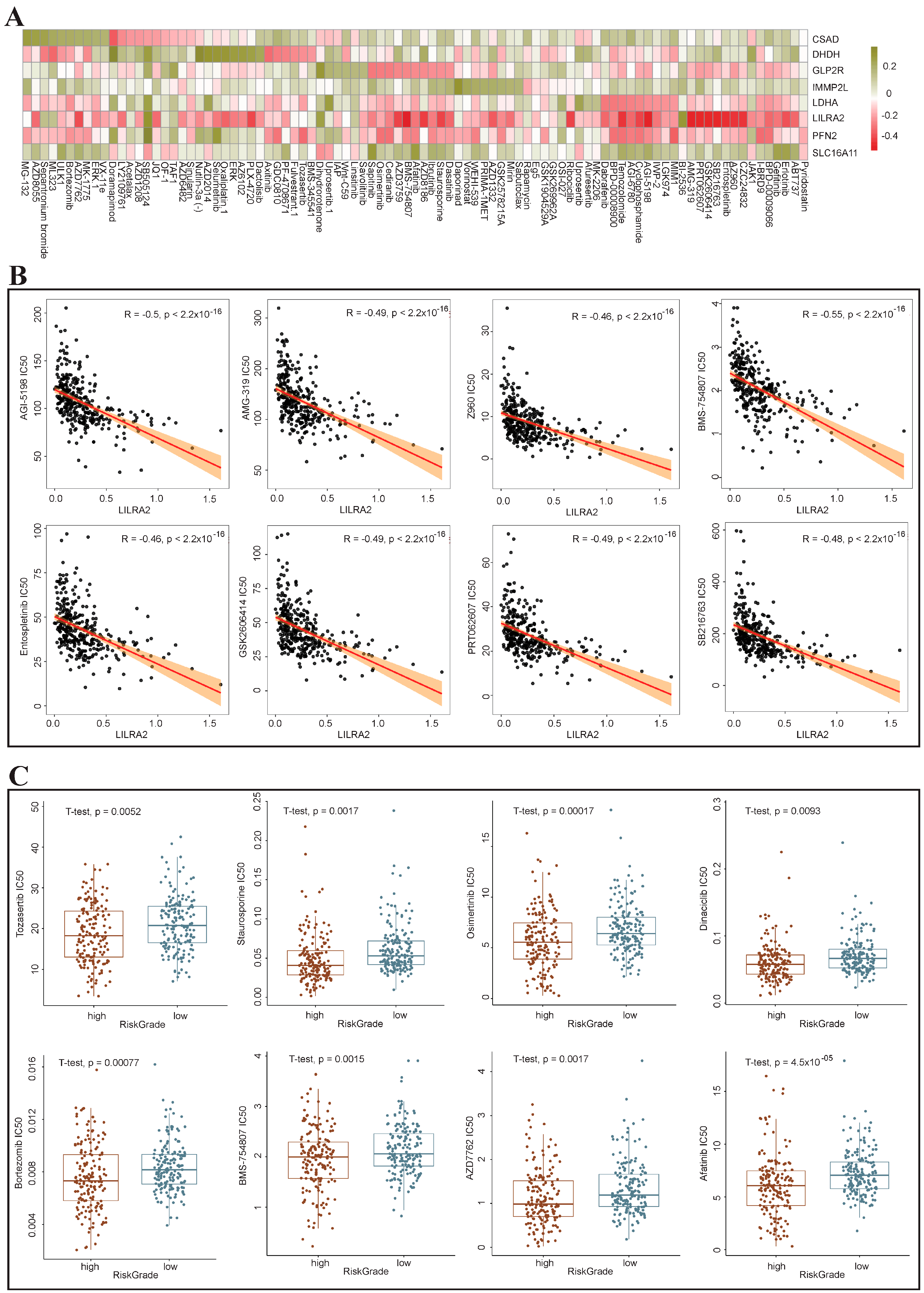
3.9. Drug Sensitivity Analysis of the High-Risk and Low-Risk Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Donne, R.; Lujambio, A. The liver cancer immune microenvironment: Therapeutic implications for hepatocellular carcinoma. Hepatology 2023, 77, 1773–1796. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Zheng, Z.; Liu, H.; Zhang, Y.; Kang, J.; Kong, X.; Rong, D.; Sun, G.; Sun, G.; Liu, L.; et al. Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biol. 2022, 56, 102463. [Google Scholar] [CrossRef]
- Xue, R.; Zhang, Q.; Cao, Q.; Kong, R.; Xiang, X.; Liu, H.; Feng, M.; Wang, F.; Cheng, J.; Li, Z.; et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature 2022, 612, 141–147. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, S.; Cai, J.; Ke, A.; Fan, J. The progress of immune checkpoint therapy in primary liver cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188638. [Google Scholar] [CrossRef]
- Xu, F.; Jin, T.; Zhu, Y.; Dai, C. Immune checkpoint therapy in liver cancer. J. Exp. Clin. Cancer Res. 2018, 37, 110. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Wong, G.; Anstee, Q.M.; Henry, L. The Global Burden of Liver Disease. Clin. Gastroenterol. Hepatol. 2023, 21, 1978–1991. [Google Scholar] [CrossRef] [PubMed]
- Calderaro, J.; Ziol, M.; Paradis, V.; Zucman-Rossi, J. Molecular and histological correlations in liver cancer. J. Hepatol. 2019, 71, 616–630. [Google Scholar] [CrossRef]
- Li, X.; Ramadori, P.; Pfister, D.; Seehawer, M.; Zender, L.; Heikenwalder, M. The immunological and metabolic landscape in primary and metastatic liver cancer. Nat. Rev. Cancer 2021, 21, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Pope, E.D., 3rd; Kimbrough, E.O.; Vemireddy, L.P.; Surapaneni, P.K.; Copland, J.A., 3rd; Mody, K. Aberrant lipid metabolism as a therapeutic target in liver cancer. Expert. Opin. Ther. Targets 2019, 23, 473–483. [Google Scholar] [CrossRef]
- Khan, M.A.; Zubair, H.; Anand, S.; Srivastava, S.K.; Singh, S.; Singh, A.P. Dysregulation of metabolic enzymes in tumor and stromal cells: Role in oncogenesis and therapeutic opportunities. Cancer Lett. 2020, 473, 176–185. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; You, C.; Meng, S.; Lai, Z.; Ai, W.; Zhang, J. EBV Infection and Its Regulated Metabolic Reprogramming in Nasopharyngeal Tumorigenesis. Front. Cell Infect. Microbiol. 2022, 12, 935205. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, X.; Wang, L.; Hong, X.; Yang, J. Metabolic reprogramming and crosstalk of cancer-related fibroblasts and immune cells in the tumor microenvironment. Front. Endocrinol. 2022, 13, 988295. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Yan, B.; Tian, X.; Liu, Q.; Jin, J.; Shi, J.; Hou, Y. CD47/SIRPalpha pathway mediates cancer immune escape and immunotherapy. Int. J. Biol. Sci. 2021, 17, 3281–3287. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Luo, Q.; Li, H.; Liu, Q.; Ju, Y.; Song, G. Increased Oxidative Phosphorylation Is Required for Stemness Maintenance in Liver Cancer Stem Cells from Hepatocellular Carcinoma Cell Line HCCLM3 Cells. Int. J. Mol. Sci. 2020, 21, 5276. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Muller, C.; Park, S.; Ryback, B.; Colombi, M.; Ritter, N.; Weissenberger, D.; Dazert, E.; Coto-Llerena, M.; Nuciforo, S.; et al. Arginine reprograms metabolism in liver cancer via RBM39. Cell 2023, 186, 5068–5083.e23. [Google Scholar] [CrossRef]
- Wu, D.; Yang, Y.; Hou, Y.; Zhao, Z.; Liang, N.; Yuan, P.; Yang, T.; Xing, J.; Li, J. Increased mitochondrial fission drives the reprogramming of fatty acid metabolism in hepatocellular carcinoma cells through suppression of Sirtuin 1. Cancer Commun 2022, 42, 37–55. [Google Scholar] [CrossRef]
- Wu, T.; Luo, G.; Lian, Q.; Sui, C.; Tang, J.; Zhu, Y.; Zheng, B.; Li, Z.; Zhang, Y.; Zhang, Y.; et al. Discovery of a Carbamoyl Phosphate Synthetase 1-Deficient HCC Subtype With Therapeutic Potential Through Integrative Genomic and Experimental Analysis. Hepatology 2021, 74, 3249–3268. [Google Scholar] [CrossRef]
- Cubuk, C.; Hidalgo, M.R.; Amadoz, A.; Rian, K.; Salavert, F.; Pujana, M.A.; Mateo, F.; Herranz, C.; Carbonell-Caballero, J.; Dopazo, J. Differential metabolic activity and discovery of therapeutic targets using summarized metabolic pathway models. NPJ Syst. Biol. Appl. 2019, 5, 7. [Google Scholar] [CrossRef]
- Maley, C.C.; Aktipis, A.; Graham, T.A.; Sottoriva, A.; Boddy, A.M.; Janiszewska, M.; Silva, A.S.; Gerlinger, M.; Yuan, Y.; Pienta, K.J.; et al. Classifying the evolutionary and ecological features of neoplasms. Nat. Rev. Cancer 2017, 17, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wu, S.; Xiong, Z.; Qu, H.; Fang, X.; Bao, Y. CROST: A comprehensive repository of spatial transcriptomics. Nucleic Acids Res. 2023, 52, D882–D890. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zheng, L.; Yoo, J.K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e1316. [Google Scholar] [CrossRef]
- Ma, L.; Heinrich, S.; Wang, L.; Keggenhoff, F.L.; Khatib, S.; Forgues, M.; Kelly, M.; Hewitt, S.M.; Saif, A.; Hernandez, J.M.; et al. Multiregional single-cell dissection of tumor and immune cells reveals stable lock-and-key features in liver cancer. Nat. Commun. 2022, 13, 7533. [Google Scholar] [CrossRef]
- Giorgi, F.M.; Ceraolo, C.; Mercatelli, D. The R Language: An Engine for Bioinformatics and Data Science. Life 2022, 12, 648. [Google Scholar] [CrossRef]
- Gribov, A.; Sill, M.; Luck, S.; Rucker, F.; Dohner, K.; Bullinger, L.; Benner, A.; Unwin, A. SEURAT: Visual analytics for the integrated analysis of microarray data. BMC Med. Genom. 2010, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, H.; Wan, L.; Wang, Z.; Wang, H.; Ge, C.; Liu, Y.; Hao, Y.; Zhang, D.; Shi, G.; et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 1118–1130. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, S.; Ma, J.; Chen, Z.; Song, G.; Rao, D.; Cheng, Y.; Huang, S.; Liu, Y.; Jiang, S.; et al. Spatiotemporal Immune Landscape of Colorectal Cancer Liver Metastasis at Single-Cell Level. Cancer Discov. 2022, 12, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Baran, J.; Cros, A.; Guberman, J.M.; Haider, S.; Hsu, J.; Liang, Y.; Rivkin, E.; Wang, J.; Whitty, B.; et al. International Cancer Genome Consortium Data Portal--a one-stop shop for cancer genomics data. Database 2011, 2011, bar026. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R.; Bien, J.; Friedman, J.; Hastie, T.; Simon, N.; Taylor, J.; Tibshirani, R.J. Strong rules for discarding predictors in lasso-type problems. J. R. Stat. Soc. Series B Stat. Methodol. 2012, 74, 245–266. [Google Scholar] [CrossRef] [PubMed]
- Blanche, P.; Dartigues, J.F.; Jacqmin-Gadda, H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat. Med. 2013, 32, 5381–5397. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 2021, 22, bbab260. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, H. Heterogeneity of liver cancer and personalized therapy. Cancer Lett. 2016, 379, 191–197. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, Z.; Li, L.; Wang, G.; Guo, L.; Luo, S.; Liao, X.; Liu, J.; Teng, W. Integrated Analysis of scRNA-Seq and Bulk RNA-Seq Reveals Metabolic Reprogramming of Liver Cancer and Establishes a Prognostic Risk Model. Genes 2024, 15, 755. https://doi.org/10.3390/genes15060755
Xiong Z, Li L, Wang G, Guo L, Luo S, Liao X, Liu J, Teng W. Integrated Analysis of scRNA-Seq and Bulk RNA-Seq Reveals Metabolic Reprogramming of Liver Cancer and Establishes a Prognostic Risk Model. Genes. 2024; 15(6):755. https://doi.org/10.3390/genes15060755
Chicago/Turabian StyleXiong, Zhuang, Lizhi Li, Guoliang Wang, Lei Guo, Shangyi Luo, Xiangwen Liao, Jingfeng Liu, and Wenhao Teng. 2024. "Integrated Analysis of scRNA-Seq and Bulk RNA-Seq Reveals Metabolic Reprogramming of Liver Cancer and Establishes a Prognostic Risk Model" Genes 15, no. 6: 755. https://doi.org/10.3390/genes15060755