
Multifactor Analyses of Frontal Cortex Lipids in the APP/PS1 Model of Familial Alzheimer’s Disease Reveal Anomalies in Responses to Dietary n-3 PUFA and Estrogenic Treatments


Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
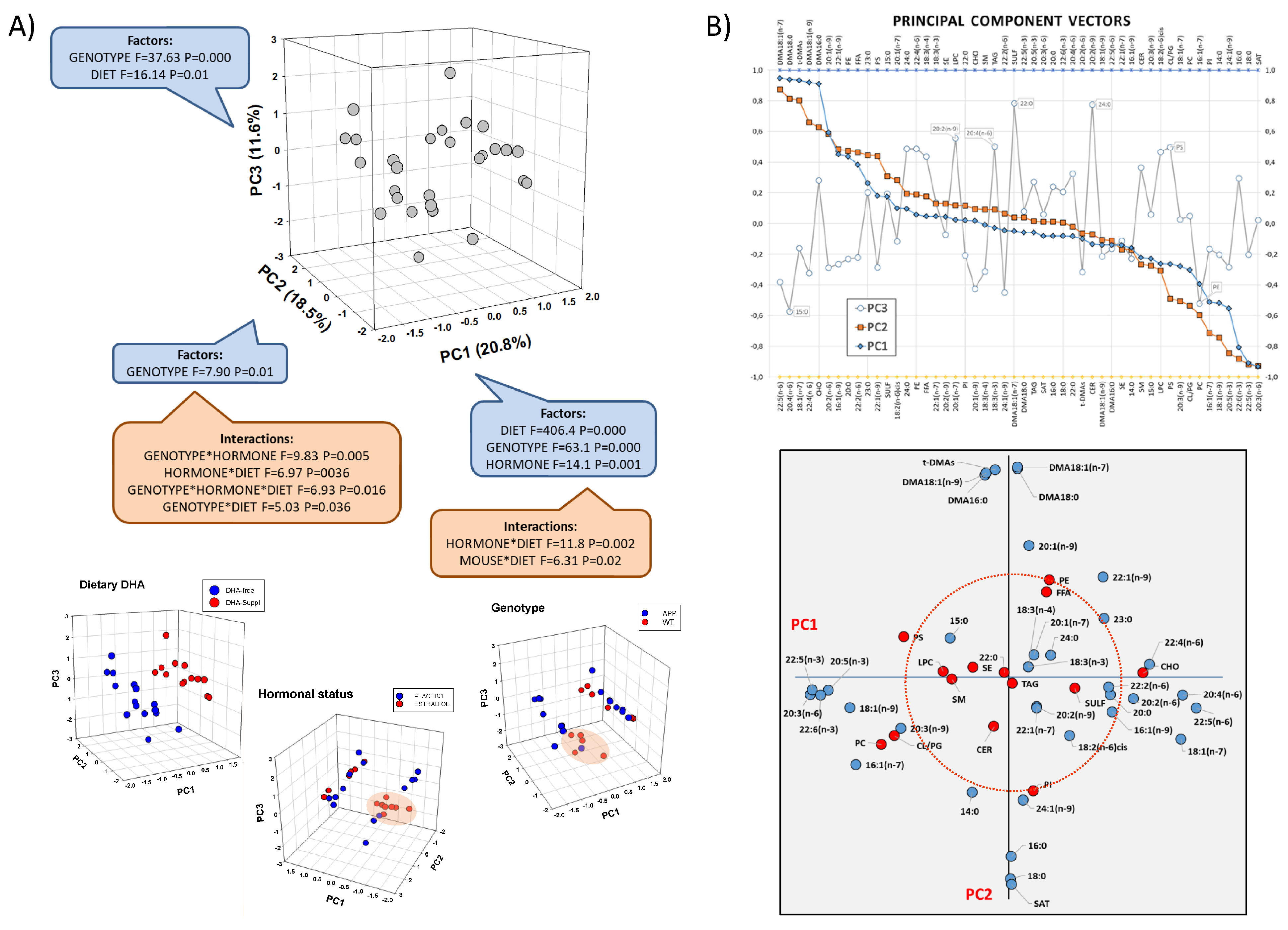
3.1. Factor Effects on the Expression of Genes Involved in LCPUFA Biosynthesis in the Frontal Cortex
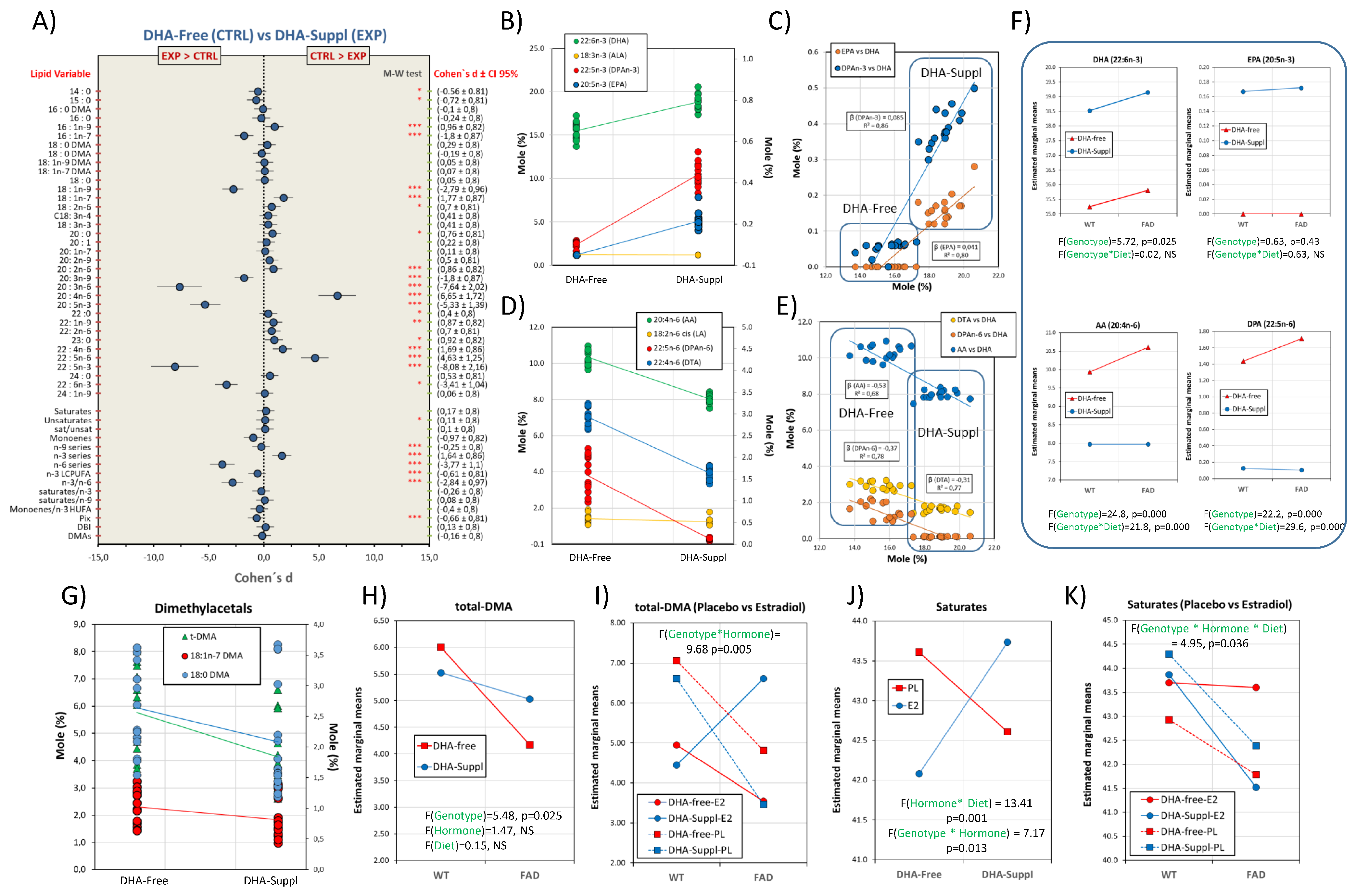
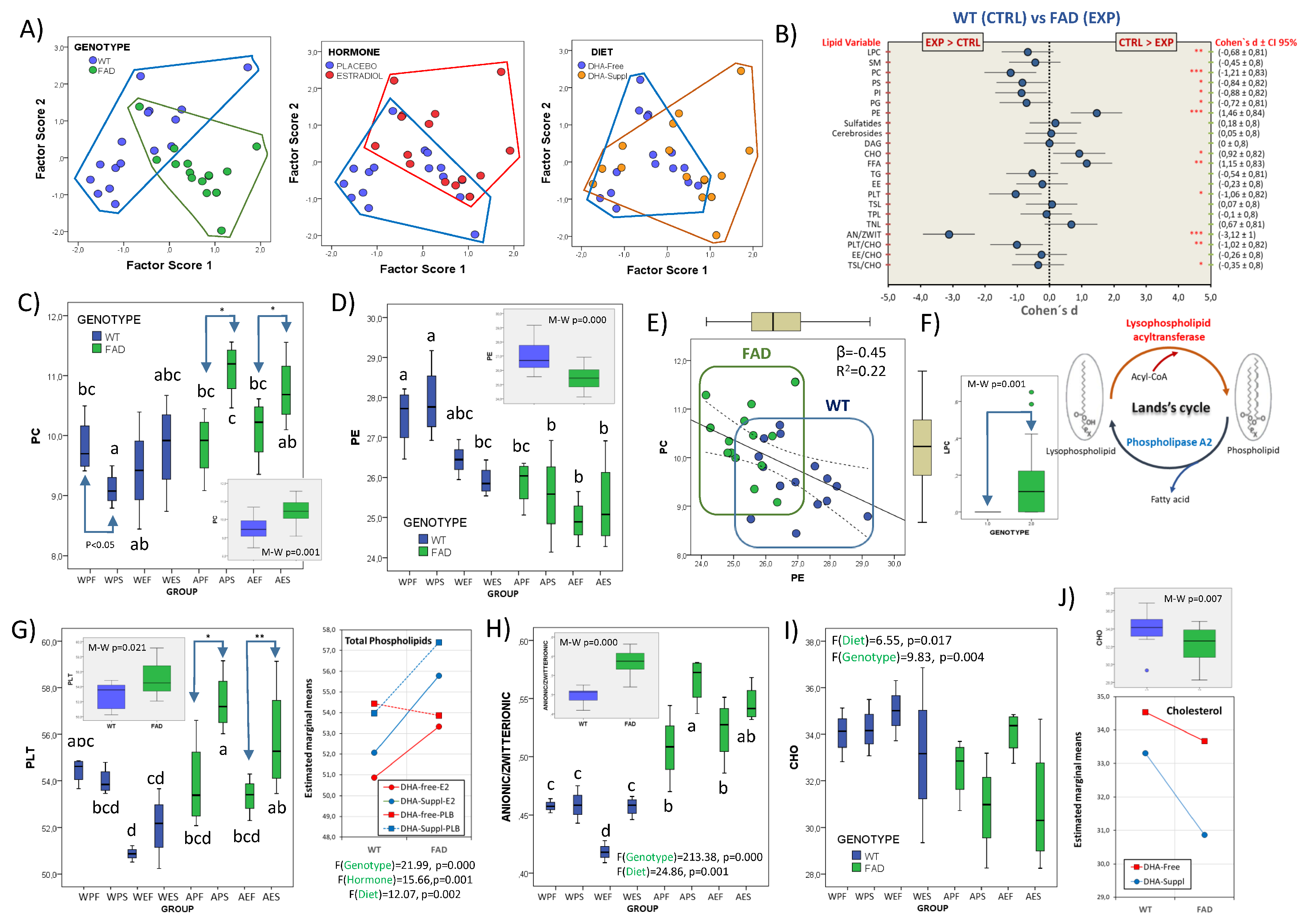
3.2. Frontal Cortex Lipid Classes Are Differentially Affected by the Presence of FAD Genotype, Circulating Estrogens, and the Dietary Condition
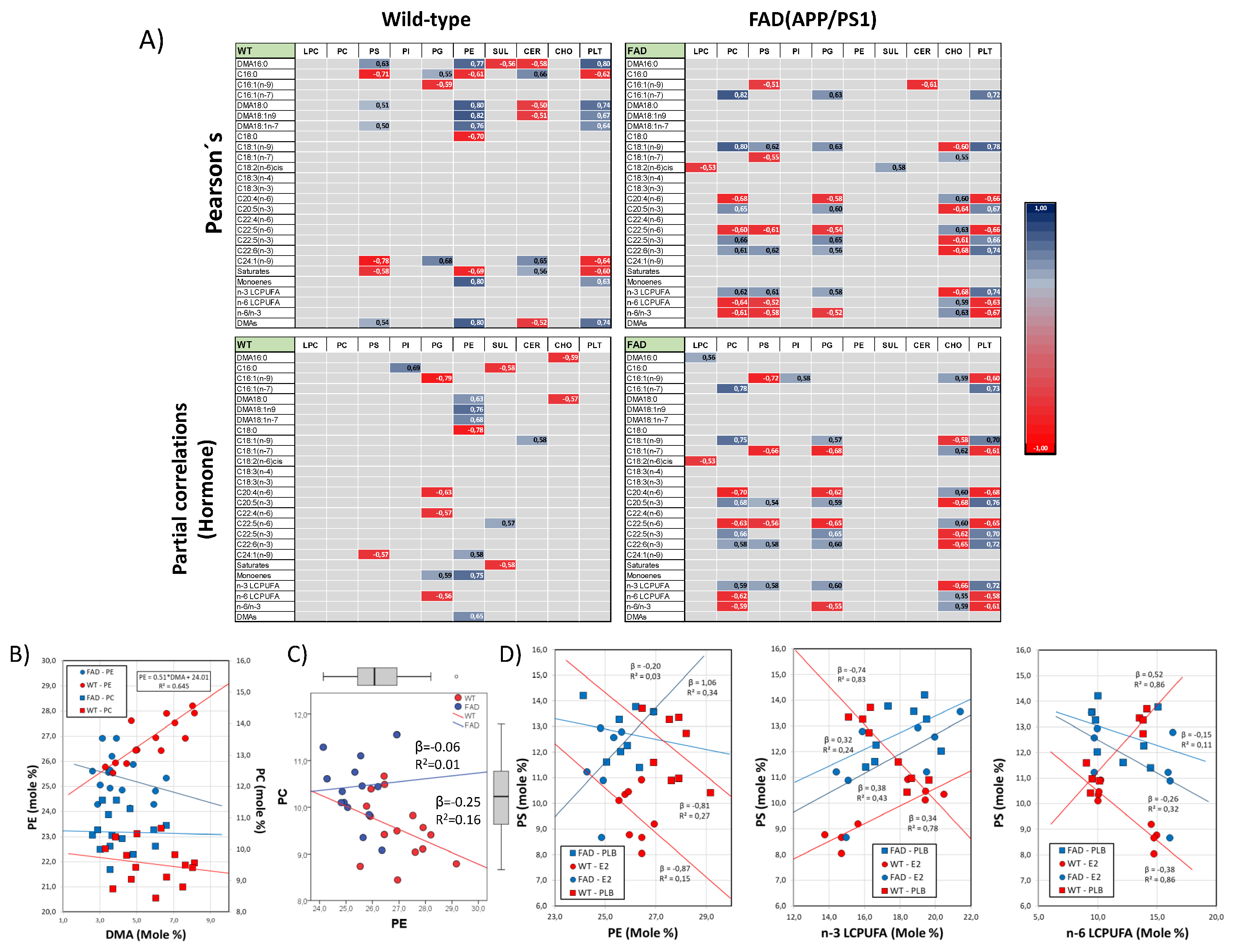
3.3. Involvement of Phospholipid Remodeling
3.4. Structural and Biophysical Correlates of Changes in Lipid Profiles
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martin, M.; Dotti, C.G.; Ledesma, M.D. Brain Cholesterol in Normal and Pathological Aging. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2010, 1801, 934–944. [Google Scholar] [CrossRef]
- Martín, V.; Fabelo, N.; Santpere, G.; Puig, B.; Marín, R.; Ferrer, I.; Díaz, M. Lipid Alterations in Lipid Rafts from Alzheimer’s Disease Human Brain Cortex. J. Alzheimer’s Dis. 2010, 19, 489–502. [Google Scholar] [CrossRef]
- Chan, R.B.; Oliveira, T.G.; Cortes, E.P.; Honig, L.S.; Duff, K.E.; Small, S.A.; Wenk, M.R.; Shui, G.; Di Paolo, G. Comparative Lipidomic Analysis of Mouse and Human Brain with Alzheimer Disease. J. Biol. Chem. 2012, 287, 2678. [Google Scholar] [CrossRef]
- Fabelo, N.; Martín, V.; Marín, R.; Moreno, D.; Ferrer, I.; Díaz, M. Altered Lipid Composition in Cortical Lipid Rafts Occurs at Early Stages of Sporadic Alzheimer’s Disease and Facilitates APP/BACE1 Interactions. Neurobiol. Aging 2014, 35, 1801–1812. [Google Scholar] [CrossRef]
- Yoon, J.H.; Seo, Y.; Jo, Y.S.; Lee, S.; Cho, E.; Cazenave-Gassiot, A.; Shin, Y.S.; Moon, M.H.; An, H.J.; Wenk, M.R.; et al. Brain Lipidomics: From Functional Landscape to Clinical Significance. Sci. Adv. 2022, 8, eadc9317. [Google Scholar] [CrossRef]
- Huang, T.L. Omega-3 Fatty Acids, Cognitive Decline, and Alzheimer’s Disease: A Critical Review and Evaluation of the Literature. J. Alzheimer’s Dis. 2010, 21, 673–690. [Google Scholar] [CrossRef]
- Astarita, G.; Jung, K.M.; Berchtold, N.C.; Nguyen, V.Q.; Gillen, D.L.; Head, E.; Cotman, C.W.; Piomelli, D. Deficient Liver Biosynthesis of Docosahexaenoic Acid Correlates with Cognitive Impairment in Alzheimer’s Disease. PLoS ONE 2010, 5, e12538. [Google Scholar] [CrossRef]
- Oster, T.; Pillot, T. Docosahexaenoic Acid and Synaptic Protection in Alzheimer’s Disease Mice. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2010, 1801, 791–798. [Google Scholar] [CrossRef]
- Moriguchi, T.; Harauma, A.; Salem, N. Plasticity of Mouse Brain Docosahexaenoic Acid: Modulation by Diet and Age. Lipids 2013, 48, 343–355. [Google Scholar] [CrossRef]
- Su, H.M. Mechanisms of N-3 Fatty Acid-Mediated Development and Maintenance of Learning Memory Performance. J. Nutr. Biochem. 2010, 21, 364–373. [Google Scholar] [CrossRef]
- Robson, L.G.; Dyall, S.C.; Sidloff, D.; Michael-Titus, A.T. Omega-3 Polyunsaturated Fatty Acids Increase the Neurite Outgrowth of Rat Sensory Neurones throughout Development and in Aged Animals. Neurobiol. Aging 2010, 31, 678–687. [Google Scholar] [CrossRef]
- Liu, J.J.; Green, P.; John Mann, J.; Rapoport, S.I.; Sublette, M.E. Pathways of Polyunsaturated Fatty Acid Utilization: Implications for Brain Function in Neuropsychiatric Health and Disease. Brain Res. 2015, 1597, 220–246. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhao, F.; Sun, Y.; Wang, Z.; Li, Q.; Wang, H.; Lu, Y. N-3 Polyunsaturated Fatty Acids in Elderly with Mild Cognitive Impairment: A Systemic Review and Meta-Analysis. J. Alzheimer’s Dis. 2024, 99, S81–S95. [Google Scholar] [CrossRef] [PubMed]
- Arevalo, M.A.; Azcoitia, I.; Garcia-Segura, L.M. The Neuroprotective Actions of Oestradiol and Oestrogen Receptors. Nat. Rev. Neurosci. 2014, 16, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Marin, R.; Guerra, B.; Alonso, R.; Ramirez, C.; Diaz, M. Estrogen Activates Classical and Alternative Mechanisms to Orchestrate Neuroprotection. Curr. Neurovasc. Res. 2005, 2, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Marin, R.; Casañas, V.; Pérez, J.A.; Fabelo, N.; Fernandez, C.E.; Diaz, M. Oestrogens as Modulators of Neuronal Signalosomes and Brain Lipid Homeostasis Related to Protection against Neurodegeneration. J. Neuroendocrinol. 2013, 25, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Craig, M.C.; Maki, P.M.; Murphy, D.G.M. The Women’s Health Initiative Memory Study: Findings and Implications for Treatment. Lancet Neurol. 2005, 4, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.L.; Zhao, J.; Li, S. Update on the Neuroprotective Effect of Estrogen Receptor Alpha Against Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 43, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M. Sex and Gender Differences in Alzheimer’s Disease Dementia. Psychiatr. Times 2018, 35, 14. [Google Scholar] [PubMed]
- Viña, J.; Lloret, A. Why Women Have More Alzheimer’s Disease than Men: Gender and Mitochondrial Toxicity of Amyloid-β Peptide. J. Alzheimer’s Dis. 2010, 20, 527–533. [Google Scholar] [CrossRef]
- Aso, E.; Lomoio, S.; Lõpez-González, I.; Joda, L.; Carmona, M.; Fernández-Yagüe, N.; Moreno, J.; Juvés, S.; Pujol, A.; Pamplona, R.; et al. Amyloid Generation and Dysfunctional Immunoproteasome Activation with Disease Progression in Animal Model of Familial Alzheimer’s Disease. Brain Pathol. 2012, 22, 636. [Google Scholar] [CrossRef]
- Götz, J.; Bodea, L.G.; Goedert, M. Rodent Models for Alzheimer Disease. Nat. Rev. Neurosci. 2018, 19, 583–598. [Google Scholar] [CrossRef]
- Xiong, H.; Callaghan, D.; Wodzinska, J.; Xu, J.; Premyslova, M.; Liu, Q.Y.; Connelly, J.; Zhang, W. Biochemical and Behavioral Characterization of the Double Transgenic Mouse Model (APPswe/PS1dE9) of Alzheimer’s Disease. Neurosci. Bull. 2011, 27, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Fabelo, N.; Martín, V.; Marín, R.; Santpere, G.; Aso, E.; Ferrer, I.; Díaz, M. Evidence for Premature Lipid Raft Aging in APP/PS1 Double-Transgenic Mice, a Model of Familial Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2012, 71, 868–881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, W.; Zan, J.; Wu, C.; Tan, W. Untargeted Lipidomics Reveals Progression of Early Alzheimer’s Disease in APP/PS1 Transgenic Mice. Sci. Rep. 2020, 10, 14509. [Google Scholar] [CrossRef] [PubMed]
- Fabelo, N.; Martin, V.; González, C.; Alonso, A.; Diaz, M. Effects of Oestradiol on Brain Lipid Class and Fatty Acid Composition: Comparison between Pregnant and Ovariectomised Oestradiol-Treated Rats. J. Neuroendocrinol. 2012, 24, 292–309. [Google Scholar] [CrossRef] [PubMed]
- Díaz, M.; Fabelo, N.; Casañas-Sánchez, V.; Marin, R.; Gómez, T.; Quinto-Alemany, D.; Pérez, J.A. Hippocampal Lipid Homeostasis in APP/PS1 Mice Is Modulated by a Complex Interplay Between Dietary DHA and Estrogens: Relevance for Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 49, 459–481. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. Statistical Power Analysis for the Behavioral Sciences; Routledge: London, UK, 1988. [Google Scholar] [CrossRef]
- Funder, D.C.; Ozer, D.J. Evaluating Effect Size in Psychological Research: Sense and Nonsense. Adv. Methods Pract. Psychol. Sci. 2019, 2, 156–168. [Google Scholar] [CrossRef]
- Plourde, M.; Cunnane, S.C. Extremely Limited Synthesis of Long Chain Polyunsaturates in Adults: Implications for Their Dietary Essentiality and Use as Supplements. Appl. Physiol. Nutr. Metab. 2007, 32, 619–634. [Google Scholar] [CrossRef]
- Brenna, J.T.; Salem, N.; Sinclair, A.J.; Cunnane, S.C. α-Linolenic Acid Supplementation and Conversion to n-3 Long-Chain Polyunsaturated Fatty Acids in Humans. Prostaglandins Leukot. Essent. Fat. Acids 2009, 80, 85–91. [Google Scholar] [CrossRef]
- Bazinet, R.P.; Layé, S. Polyunsaturated Fatty Acids and Their Metabolites in Brain Function and Disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Barceló-Coblijn, G.; Murphy, E.J. Alpha-Linolenic Acid and Its Conversion to Longer Chain N−3 Fatty Acids: Benefits for Human Health and a Role in Maintaining Tissue N−3 Fatty Acid Levels. Prog. Lipid Res. 2009, 48, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, S.I.; Rao, J.S.; Igarashi, M. Brain Metabolism of Nutritionally Essential Polyunsaturated Fatty Acids Depends on Both the Diet and the Liver. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, S.I.; Igarashi, M. Can the Rat Liver Maintain Normal Brain DHA Metabolism in the Absence of Dietary DHA? Prostaglandins Leukot. Essent. Fat. Acids 2009, 81, 119–123. [Google Scholar] [CrossRef]
- Kim, H.W.; Rao, J.S.; Rapoport, S.I.; Igarashi, M. Regulation of Rat Brain Polyunsaturated Fatty Acid (PUFA) Metabolism during Graded Dietary n-3 PUFA Deprivation. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Bourre, J.M.; Dumont, O.; Pascal, G.; Durand, G. Dietary α-Linolenic Acid at 1.3 g/Kg Maintains Maximal Docosahexaenoic Acid Concentration in Brain, Heart and Liver of Adult Rats. J. Nutr. 1993, 123, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Taha, A.Y.; Chang, L.; Chen, M. Threshold Changes in Rat Brain Docosahexaenoic Acid Incorporation and Concentration Following Graded Reductions in Dietary Alpha-Linolenic Acid. Prostaglandins Leukot. Essent. Fat. Acids 2016, 105, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.C.; Ho, P.C.; Tu, Y.K.; Jou, I.M.; Tsai, K.J. Lipids and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1505. [Google Scholar] [CrossRef]
- Ciappolino, V.; Mazzocchi, A.; Botturi, A.; Turolo, S.; Delvecchio, G.; Agostoni, C.; Brambilla, P. The Role of Docosahexaenoic Acid (DHA) on Cognitive Functions in Psychiatric Disorders. Nutrients 2019, 11, 769. [Google Scholar] [CrossRef]
- Joffre, C.; Nadjar, A.; Lebbadi, M.; Calon, F.; Laye, S. N-3 LCPUFA Improves Cognition: The Young, the Old and the Sick. Prostaglandins Leukot. Essent. Fat. Acids 2014, 91, 1–20. [Google Scholar] [CrossRef]
- Brossard, N.; Croset, M.; Pachiaudi, C.; Riou, J.P.; Tayot, J.L.; Lagarde, M. Retroconversion and Metabolism of [13C]22:6n−3 in Humans and Rats after Intake of a Single Dose of [13C]22:6n−3-Triacylglycerols. Am. J. Clin. Nutr. 1996, 64, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Metherel, A.H.; Bazinet, R.P. Updates to the N-3 Polyunsaturated Fatty Acid Biosynthesis Pathway: DHA Synthesis Rates, Tetracosahexaenoic Acid and (Minimal) Retroconversion. Prog. Lipid Res. 2019, 76, 101008. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; DeMar, J.C.; Ma, K.; Chang, L.; Bell, J.M.; Rapoport, S.I. Docosahexaenoic Acid Synthesis from α-Linolenic Acid by Rat Brain Is Unaffected by Dietary n-3 PUFA Deprivation. J. Lipid Res. 2007, 48, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Kochman, K.; Czauderna, M. The Necessity of Adequate Nutrition with Diets Containing Omega-3 and Omega-6 Fatty Acids for Proper Brain Development, Function and Delayed Aging: Review. J. Anim. Feed Sci. 2010, 19, 511–524. [Google Scholar] [CrossRef]
- Francis, H.; Stevenson, R. The Longer-Term Impacts of Western Diet on Human Cognition and the Brain. Appetite 2013, 63, 119–128. [Google Scholar] [CrossRef]
- Janssen, C.I.F.; Kiliaan, A.J. Long-Chain Polyunsaturated Fatty Acids (LCPUFA) from Genesis to Senescence: The Influence of LCPUFA on Neural Development, Aging, and Neurodegeneration. Prog. Lipid Res. 2014, 53, 1–17. [Google Scholar] [CrossRef]
- Diaz, M.; Marin, R. Brain Polyunsaturated Lipids and Neurodegenerative Diseases. In Nutraceuticals and Functional Foods: Natural Remedy; Nova Science Pub Inc.: Hauppauge, NY, USA, 2014; pp. 387–412. [Google Scholar]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism Pathways of Arachidonic Acids: Mechanisms and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef]
- Igarashi, M.; Kim, H.W.; Gao, F.; Chang, L.; Ma, K.; Rapoport, S.I. Fifteen Weeks of Dietary N-3 Polyunsaturated Fatty Acid Deprivation Increase Turnover of n-6 Docosapentaenoic Acid in Rat-Brain Phospholipids. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2012, 1821, 1235–1243. [Google Scholar] [CrossRef]
- Axelsen, P.H.; Murphy, R.C.; Igarashi, M.; Rapoport, S.I. Increased Ω6-Containing Phospholipids and Primary Ω6 Oxidation Products in the Brain Tissue of Rats on an Ω3-Deficient Diet. PLoS ONE 2016, 11, e0164326. [Google Scholar] [CrossRef]
- Yokoyama, M.; Kobayashi, H.; Tatsumi, L.; Tomita, T. Mouse Models of Alzheimer’s Disease. Front. Mol. Neurosci. 2022, 15, 912995. [Google Scholar] [CrossRef]
- Zhong, M.Z.; Peng, T.; Duarte, M.L.; Wang, M.; Cai, D. Updates on Mouse Models of Alzheimer’s Disease. Mol. Neurodegener. 2024, 19, 23. [Google Scholar] [CrossRef]
- Ginsberg, L.; Rafique, S.; Xuereb, J.H.; Rapoport, S.I.; Gershfeld, N.L. Disease and Anatomic Specificity of Ethanolamine Plasmalogen Deficiency in Alzheimer’s Disease Brain. Brain Res. 1995, 698, 223–226. [Google Scholar] [CrossRef]
- Naudí, A.; Cabré, R.; Jové, M.; Ayala, V.; Gonzalo, H.; Portero-Otín, M.; Ferrer, I.; Pamplona, R. Lipidomics of Human Brain Aging and Alzheimer’s Disease Pathology. Int. Rev. Neurobiol. 2015, 122, 133–189. [Google Scholar] [CrossRef]
- Wood, P.L.; Barnette, B.L.; Kaye, J.A.; Quinn, J.F.; Woltjer, R.L. Non-Targeted Lipidomics of CSF and Frontal Cortex Grey and White Matter in Control, Mild Cognitive Impairment, and Alzheimer’s Disease Subjects. Acta Neuropsychiatr. 2015, 27, 270–278. [Google Scholar] [CrossRef]
- Morselli, E.; de Souza Santos, R.; Gao, S.; Ávalos, Y.; Criollo, A.; Palmer, B.F.; Clegg, D.J. Impact of Estrogens and Estrogen Receptor-α in Brain Lipid Metabolism. Am. J. Physiol.-Endocrinol. Metab. 2018, 315, E7–E14. [Google Scholar] [CrossRef] [PubMed]
- Cuenca-Bermejo, L.; Prinetti, A.; Kublickiene, K.; Raparelli, V.; Kautzky-Willer, A.; Norris, C.M.; Pilote, L.; Herrero, M.T. Fundamental Neurochemistry Review: Old Brain Stories-Influence of Age and Sex on the Neurodegeneration-Associated Lipid Changes. J. Neurochem. 2023, 166, 427–452. [Google Scholar] [CrossRef] [PubMed]
- Marin, R.; Diaz, M. Estrogen Interactions with Lipid Rafts Related to Neuroprotection. Impact of Brain Ageing and Menopause. Front. Neurosci. 2018, 12, 328334. [Google Scholar] [CrossRef] [PubMed]
- Guillou, H.; Zadravec, D.; Martin, P.G.P.; Jacobsson, A. The Key Roles of Elongases and Desaturases in Mammalian Fatty Acid Metabolism: Insights from Transgenic Mice. Prog. Lipid Res. 2010, 49, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Kent, C. Eukaryotic Phospholipid Biosynthesis. Annu. Rev. Biochem. 1995, 64, 315–343. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, F.; Smith, T.K. The Kennedy Pathway—De Novo Synthesis of Phosphatidylethanolamine and Phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tontonoz, P. Phospholipid Remodeling in Physiology and Disease. Annu. Rev. Physiol. 2019, 81, 165–188. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Boucrot, E. Membrane Curvature at a Glance. J. Cell Sci. 2015, 128, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Aimon, S.; Callan-Jones, A.; Berthaud, A.; Pinot, M.; Toombes, G.E.S.; Bassereau, P. Membrane Shape Modulates Transmembrane Protein Distribution. Dev. Cell 2014, 28, 212. [Google Scholar] [CrossRef] [PubMed]
- Bigay, J.; Antonny, B. Curvature, Lipid Packing, and Electrostatics of Membrane Organelles: Defining Cellular Territories in Determining Specificity. Dev. Cell 2012, 23, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Terakawa, M.S.; Lin, Y.; Kinoshita, M.; Kanemura, S.; Itoh, D.; Sugiki, T.; Okumura, M.; Ramamoorthy, A.; Lee, Y.H. Impact of Membrane Curvature on Amyloid Aggregation. Biochim. Biophys. Acta-Biomembr. 2018, 1860, 1741–1764. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, Y.; Ikeda, K.; Nakano, M. High Membrane Curvature Enhances Binding, Conformational Changes, and Fibrillation of Amyloid-β on Lipid Bilayer Surfaces. Langmuir 2015, 31, 11549–11557. [Google Scholar] [CrossRef] [PubMed]
- Cantor, R.S. Lateral Pressures in Cell Membranes: A Mechanism for Modulation of Protein Function. J. Phys. Chem. B 1997, 101, 1723–1725. [Google Scholar] [CrossRef]
- Diaz, M.L.; Fabelo, N.; Marín, R. Genotype-Induced Changes in Biophysical Properties of Frontal Cortex Lipid Raft from APP/PS1 Transgenic Mice. Front. Physiol. 2012, 3, 37031. [Google Scholar] [CrossRef]
- Soni, S.P.; LoCascio, D.S.; Liu, Y.; Williams, J.A.; Bittman, R.; Stillwell, W.; Wassall, S.R. Docosahexaenoic Acid Enhances Segregation of Lipids between: 2H-NMR Study. Biophys. J. 2008, 95, 203–214. [Google Scholar] [CrossRef]
- Díaz, M.; Pereda de Pablo, D.; Valdés-Baizabal, C.; Santos, G.; Marin, R. Molecular and Biophysical Features of Hippocampal “Lipid Rafts Aging” Are Modified by Dietary n-3 Long-Chain Polyunsaturated Fatty Acids. Aging Cell 2023, 22, e13867. [Google Scholar] [CrossRef]
- Díaz, M.; Valdés-Baizabal, C.; de Pablo, D.P.; Marin, R. Age-Dependent Changes in Nrf2/Keap1 and Target Antioxidant Protein Expression Correlate to Lipoxidative Adducts, and Are Modulated by Dietary N-3 LCPUFA in the Hippocampus of Mice. Antioxidants 2024, 13, 206. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | WT | FAD | FAD | |||||||||||
| PLACEBO | E2 | PLACEBO | E2 | |||||||||||
| DHA-Free vs. DHA-Suppl | DHA-Free vs. DHA-Suppl | DHA-Free vs. DHA-Suppl | DHA-Free vs. DHA-Suppl | |||||||||||
| Gene | Expression | p-Value | Gene | Expression | p-Value | Gene | Expression | p-Value | Gene | Expression | p-Value | |||
| Elovl2 | 0.974 | 0.833 | Elovl2 | 0.953 | 0.769 | Elovl2 | 0.277 | 0.067 | Elovl2 | 0.661 | 0.067 | |||
| Elovl4 | 0.988 | 0.887 | Elovl4 | 0.935 | 0.61 | Elovl4 | 0.273 | 0.056 | Elovl4 | 0.66 | 0.032 | |||
| Elovl5 | 0.803 | 0.698 | Elovl5 | 1.438 | 0.571 | Elovl5 | 0.054 | 0.063 | Elovl5 | 0.707 | 0.042 | |||
| Scd1 | 1.521 | 0.079 | Scd1 | 1.074 | 0.536 | Scd1 | 0.554 | 0.067 | Scd1 | 0.741 | 0.137 | |||
| Scd2 | 1.143 | 0.131 | Scd2 | 1.116 | 0.571 | Scd2 | 0.334 | 0.029 | Scd2 | 0.749 | 0.013 | |||
| Fads1 | 0.782 | 0.388 | Fads1 | 1.479 | 0.343 | Fads1 | 0.057 | 0.029 | Fads1 | 0.731 | 0.053 | |||
| Fads2 | 0.959 | 0.516 | Fads2 | 1.123 | 0.287 | Fads2 | 0.923 | 0.759 | Fads2 | 0.903 | 0.426 | |||
| PLACEBO | PLACEBO | E2 | E2 | |||||||||||
| DHA-Free | DHA-Suppl | DHA-Free | DHA-Suppl | |||||||||||
| WT vs. FAD | WT vs. FAD | WT vs. FAD | WT vs. FAD | |||||||||||
| Gene | Expression | p-Value | Gene | Expression | p-Value | Gene | Expression | p-Value | Gene | Expression | P-Value | |||
| Acox1 | 1.416 | 0.033 | Acox1 | 0.184 | 0.034 | Acox1 | 1.473 | 0.137 | Acox1 | 1.07 | 0.443 | |||
| Elovl2 | 1.917 | 0.036 | Elovl2 | 0.542 | 0.173 | Elovl2 | 1.826 | 0.026 | Elovl2 | 1.266 | 0.275 | |||
| Elovl4 | 1.467 | 0.029 | Elovl4 | 0.397 | 0.012 | Elovl4 | 1.596 | 0.032 | Elovl4 | 1.117 | 0.138 | |||
| Elovl5 | 1.607 | 0.053 | Elovl5 | 0.105 | 0.078 | Elovl5 | 2.113 | 0.238 | Elovl5 | 1.038 | 0.741 | |||
| Scd1 | 1.986 | 0.068 | Scd1 | 0.723 | 0.285 | Scd1 | 1.6 | 0.000 | Scd1 | 1.102 | 0.433 | |||
| Scd2 | 1.635 | 0.053 | Scd2 | 0.478 | 0.096 | Scd2 | 1.599 | 0.054 | Scd2 | 1.085 | 0.465 | |||
| Fads1 | 1.069 | 0.540 | Fads1 | 0.079 | 0.054 | Fads1 | 1.434 | 0.425 | Fads1 | 0.708 | 0.024 | |||
| Fads2 | 1.211 | 0.154 | Fads2 | 1.162 | 0.509 | Fads2 | 1.299 | 0.188 | Fads2 | 1.041 | 0.684 | |||
| WT | WT | FAD | FAD | |||||||||||
| DHA-Free | DHA-Suppl | DHA-Free | DHA-Suppl | |||||||||||
| PLACEBO vs. E2 | PLACEBO vs. E2 | PLACEBO vs. E2 | PLACEBO vs. E2 | |||||||||||
| Gene | Expression | p-Value | Gene | Expression | p-Value | Gene | Expression | p-Value | Gene | Expression | p-Value | |||
| Acox1 | 0.958 | 0.898 | Acox1 | 1.091 | 0.801 | Acox1 | 1.004 | 1.000 | Acox1 | 6.20 | 0.088 | |||
| Elovl2 | 1.107 | 0.884 | Elovl2 | 1.086 | 0.841 | Elovl2 | 1.042 | 0.755 | Elovl2 | 2.51 | 0.061 | |||
| Elovl4 | 0.975 | 0.793 | Elovl4 | 0.927 | 0.593 | Elovl4 | 0.992 | 0.967 | Elovl4 | 2.51 | 0.081 | |||
| Elovl5 | 0.788 | 0.741 | Elovl5 | 1.412 | 0.809 | Elovl5 | 1.044 | 0.735 | Elovl5 | 13.98 | 0.082 | |||
| Scd1 | 1.231 | 0.299 | Scd1 | 0.868 | 0.254 | Scd1 | 0.991 | 0.967 | Scd1 | 1.32 | 0.384 | |||
| Scd2 | 1.032 | 0.798 | Scd2 | 1.01 | 0.869 | Scd2 | 1.012 | 0.934 | Scd2 | 2.32 | 0.09 | |||
| Fads1 | 0.794 | 0.586 | Fads1 | 1.499 | 0.022 | Fads1 | 1.073 | 0.447 | Fads1 | 13.45 | 0.061 | |||
| Fads2 | 0.946 | 0.459 | Fads2 | 1.107 | 0.214 | Fads2 | 1.01 | 0.846 | Fads2 | 0.98 | 0.901 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz, M. Multifactor Analyses of Frontal Cortex Lipids in the APP/PS1 Model of Familial Alzheimer’s Disease Reveal Anomalies in Responses to Dietary n-3 PUFA and Estrogenic Treatments. Genes 2024, 15, 810. https://doi.org/10.3390/genes15060810
Díaz M. Multifactor Analyses of Frontal Cortex Lipids in the APP/PS1 Model of Familial Alzheimer’s Disease Reveal Anomalies in Responses to Dietary n-3 PUFA and Estrogenic Treatments. Genes. 2024; 15(6):810. https://doi.org/10.3390/genes15060810
Chicago/Turabian StyleDíaz, Mario. 2024. "Multifactor Analyses of Frontal Cortex Lipids in the APP/PS1 Model of Familial Alzheimer’s Disease Reveal Anomalies in Responses to Dietary n-3 PUFA and Estrogenic Treatments" Genes 15, no. 6: 810. https://doi.org/10.3390/genes15060810
APA StyleDíaz, M. (2024). Multifactor Analyses of Frontal Cortex Lipids in the APP/PS1 Model of Familial Alzheimer’s Disease Reveal Anomalies in Responses to Dietary n-3 PUFA and Estrogenic Treatments. Genes, 15(6), 810. https://doi.org/10.3390/genes15060810