
From Alpha-Thalassemia Trait to NPRL3-Related Epilepsy: A Genomic Diagnostic Odyssey
 ,
,
Abstract
:1. Introduction
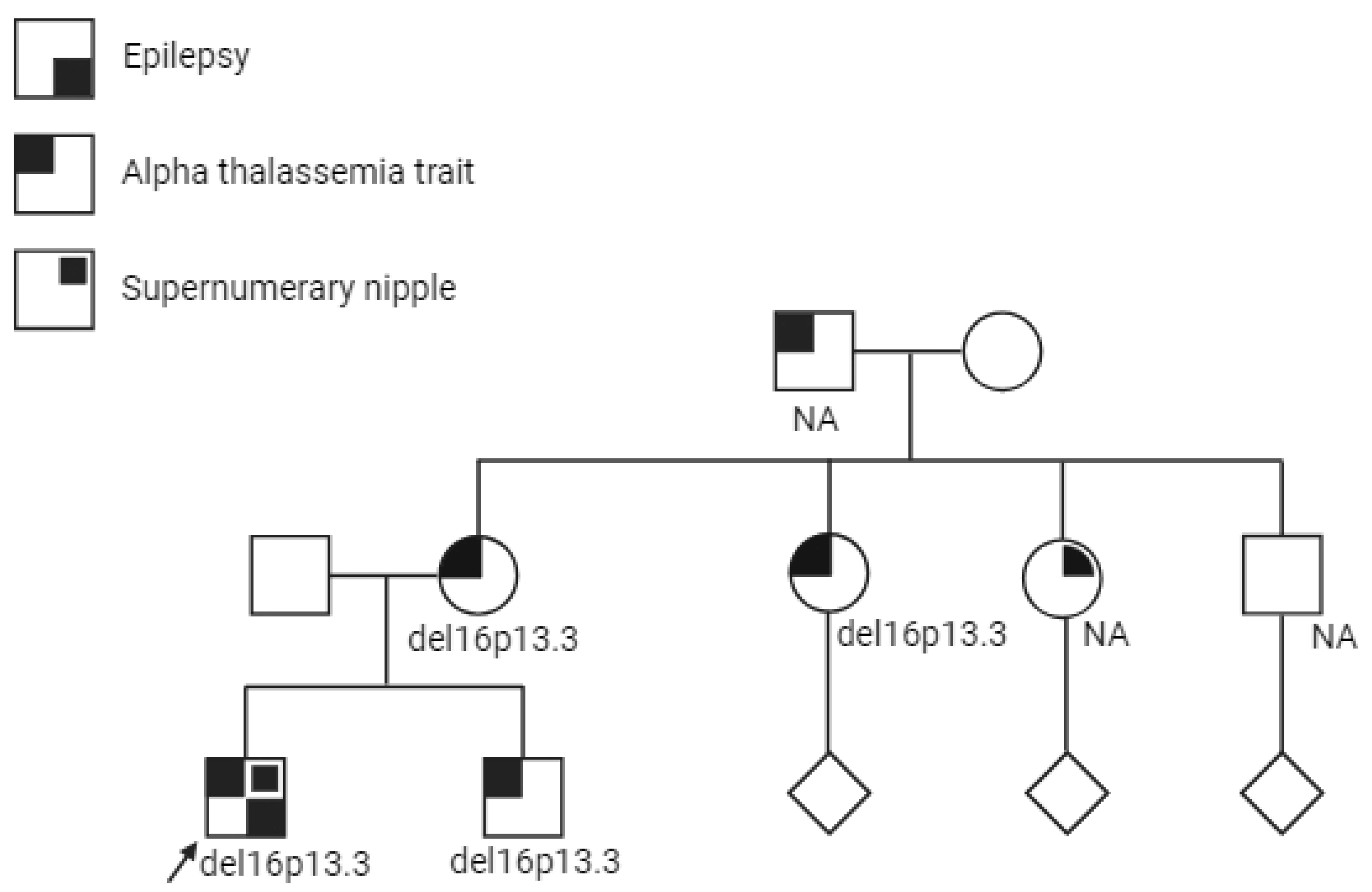
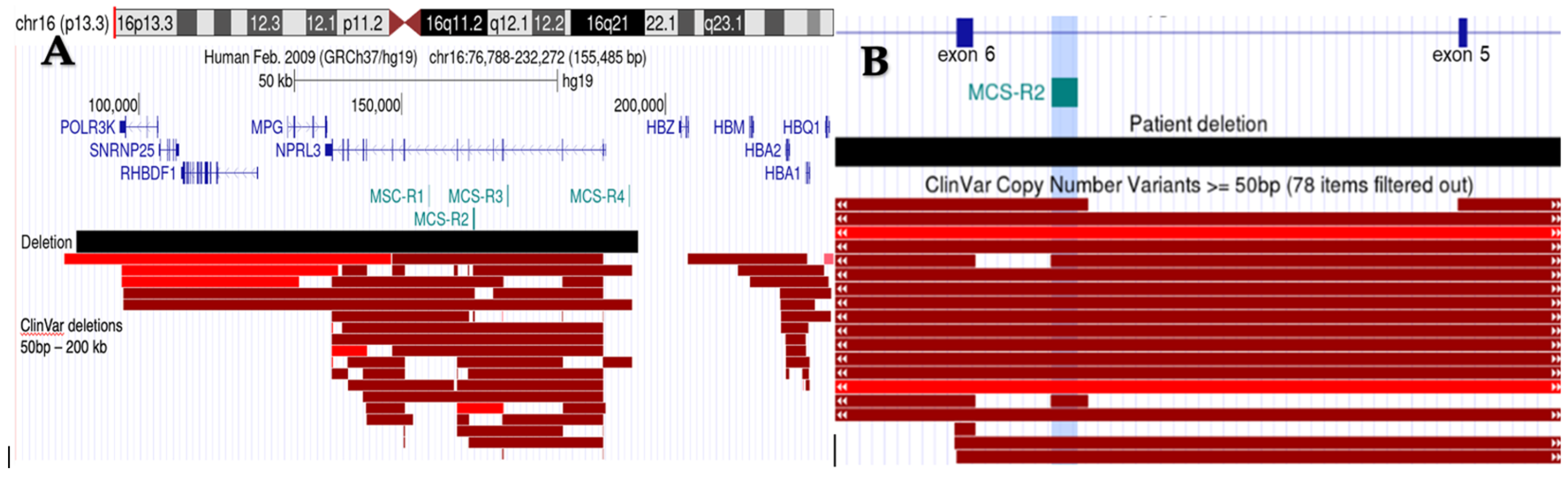
2. Case Presentation
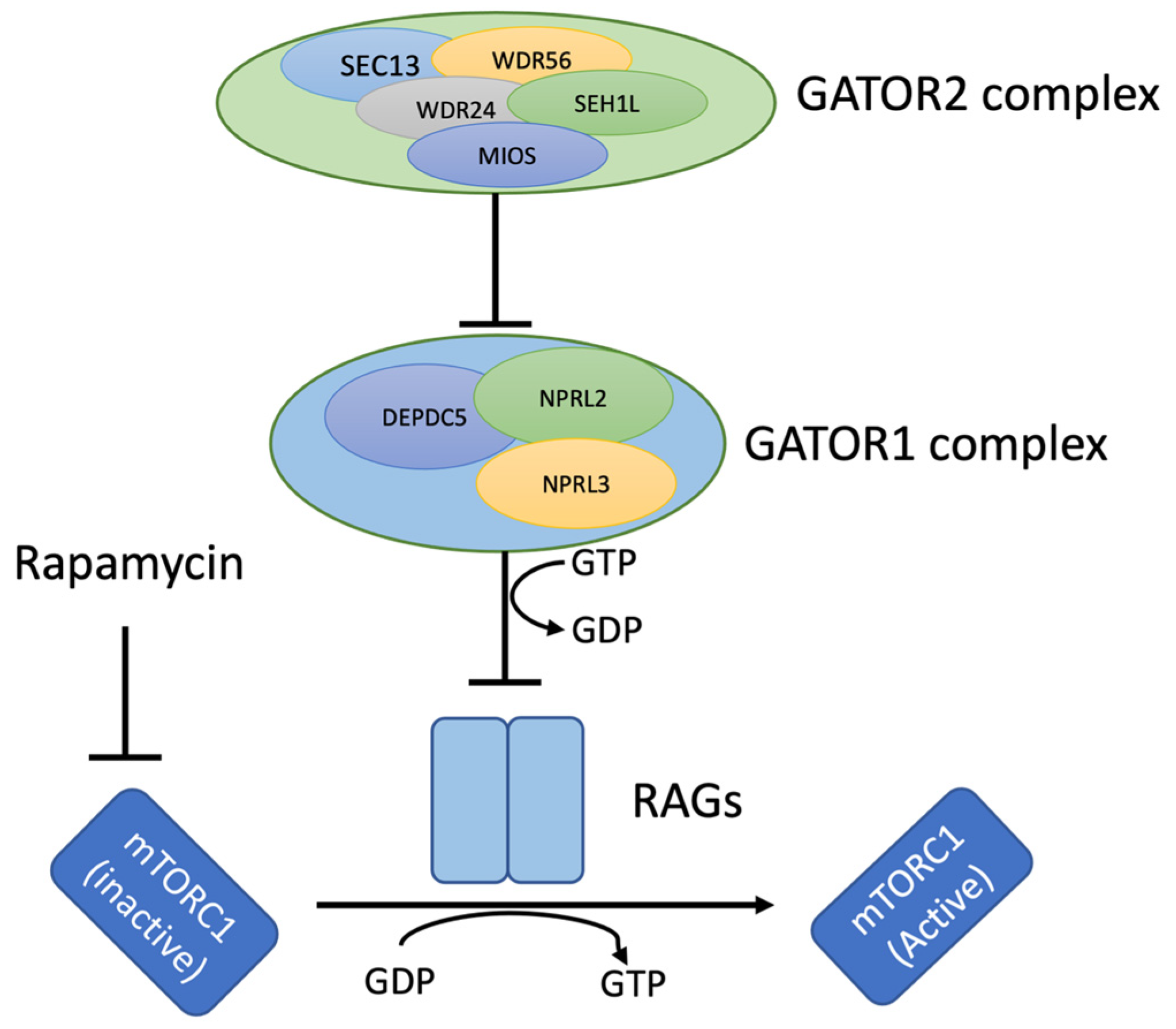
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, H.; Deng, J.; Wang, X.; Chen, C.; Chen, S.; Dai, L.; Fang, F. Clinical phenotypic and genotypic characterization of NPRL3-related epilepsy. Front. Neurol. 2023, 14, 1113747. [Google Scholar] [CrossRef] [PubMed]
- Weckhuysen, S.; Marsan, E.; Lambrecq, V.; Marchal, C.; Morin-Brureau, M.; An-Gourfinkel, I.; Baulac, M.; Fohlen, M.; Kallay Zetchi, C.; Seeck, M.; et al. Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia 2016, 57, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, X.; Wang, S.; Xu, K.; Huang, S.; Zhu, S. A Novel Loss-of-Function Mutation in the NPRL3 Gene Identified in Chinese Familial Focal Epilepsy with Variable Foci. Front. Genet. 2021, 12, 766354. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Gao, X.; Chen, L.; Kan, Y.; Du, Z.; Xin, S.; Ji, W.; Yu, Q.; Cao, L. Identification of two rare NPRL3 variants in two Chinese families with familial focal epilepsy with variable foci 3: NGS analysis with literature review. Front. Genet. 2022, 13, 1054567. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, A.; Antel, S.B.; Collins, D.L.; Bernasconi, N.; Olivier, A.; Dubeau, F.; Pike, G.B.; Andermann, F.; Arnold, D.L. Texture analysis and morphological processing of magnetic resonance imaging assist detection of focal cortical dysplasia in extra-temporal partial epilepsy. Ann. Neurol. 2001, 49, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Wang, J.; Qin, Z.; Feng, J.; Mao, C.; Chen, Y.; Huang, X.; Ruan, Y. Phenotypic and genotypic characterization of NPRL3-related epilepsy: Two case reports and literature review. Epilepsia Open 2024, 9, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Zeng, S.; Song, L.; Ma, H.; Chen, R.; Luo, J.; Wang, X.; Ma, T.; Xu, X.; Sun, H.; et al. Functional characterization of novel NPRL3 mutations identified in three families with focal epilepsy. Sci. China Life Sci. 2023, 66, 2152–2166. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.E.; Nieto Guil, A.F.; Robertson, L.J.; Piltz, S.G.; Hughes, J.N.; Thomas, P.Q. Functional screening of GATOR1 complex variants reveals a role for mTORC1 deregulation in FCD and focal epilepsy. Neurobiol. Dis. 2020, 134, 104640. [Google Scholar] [CrossRef] [PubMed]
- Iffland, P.H.; Everett, M.E.; Cobb-Pitstick, K.M.; Bowser, L.E.; Barnes, A.E.; Babus, J.K.; Romanowski, A.J.; Baybis, M.; Elziny, S.; Puffenberger, E.G.; et al. NPRL3 loss alters neuronal morphology, mTOR localization, cortical lamination and seizure threshold. Brain 2022, 145, 3872–3885. [Google Scholar] [CrossRef]
- Dainelli, A.; Iacomino, M.; Rossato, S.; Bugin, S.; Traverso, M.; Severino, M.; Gustincich, S.; Capra, V.; Di Duca, M.; Zara, F.; et al. Refining the electroclinical spectrum of NPRL3-related epilepsy: A novel multiplex family and literature review. Epilepsia Open 2023, 8, 1314–1330. [Google Scholar] [CrossRef]
- Vawter-Lee, M.; Franz, D.N.; Fuller, C.E.; Greiner, H.M. Clinical Letter: A case report of targeted therapy with sirolimus for NPRL3 epilepsy. Seizure 2019, 73, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Bernet, A.; Sabatier, S.; Picketts, D.J.; Ouazana, R.; Morle, F.; Higgs, D.R.; Godet, J. Targeted inactivation of the major positive regulatory element (HS-40) of the human α-globin gene locus. Blood 1995, 86, 1202–1211. [Google Scholar] [CrossRef]
- Sharpe, J.A.; Wells, D.J.; Whitelaw, E.; Vyas, P.; Higgs, D.R.; Wood, W.G. Analysis of the human α-globin gene cluster in transgenic mice. Proc. Natl. Acad. Sci. USA 1993, 90, 11262–11266. [Google Scholar] [CrossRef] [PubMed]
- Vernimmen, D.; Marques-Kranc, F.; Sharpe, J.A.; Sloane-Stanley, J.A.; Wood, W.G.; Wallace, H.A.; Smith, A.J.; Higgs, D.R. Chromosome looping at the human α-globin locus is mediated via the major upstream regulatory element (HS–40). Blood 2009, 114, 4253–4260. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.; Picanço, I.; Seuanes, F.; Seixas, M.T.; Faustino, P. Novel large deletions in the human α-globin gene cluster: Clarifying the HS-40 long-range regulatory role in the native chromosome environment. Blood Cells Mol. Dis. 2010, 45, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Stephenson, S.E.; Pope, K.; Gillies, G.; Maixner, W.; Macdonald-Laurs, E.; MacGregor, D.; D’Arcy, C.; Jackson, G.; Harvey, A.S.; et al. Genetic characterization identifies bottom-of-sulcus dysplasia as an mTORopathy. Neurology 2020, 95, e2542–e2551. [Google Scholar] [CrossRef] [PubMed]
- Sahly, A.N.; Whitney, R.; Costain, G.; Chau, V.; Otsubo, H.; Ochi, A.; Donner, E.J.; Cunningham, J.; Jones, K.C.; Widjaja, E.; et al. Epilepsy surgery outcomes in patients with GATOR1 gene complex variants: Report of new cases and review of literature. Seizure 2023, 107, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Heron, S.E.; Regan, B.M.; Mandelstam, S.; Crompton, D.E.; Hodgson, B.L.; Licchetta, L.; Provini, F.; Bisulli, F.; Vadlamudi, L.; et al. Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann. Neurol. 2014, 75, 782–787. [Google Scholar] [CrossRef]
- Abumurad, S.; Issa, N.P.; Wu, S.; Rose, S.; Esengul, Y.T.; Nordli, D.; Warnke, P.C.; Tao, J.X. Laser interstitial thermal therapy for NPRL3-related epilepsy with multiple seizure foci: A case report. Epilepsy Behav. Rep. 2021, 16, 100459. [Google Scholar] [CrossRef]
- Sim, J.C.; Scerri, T.; Fanjul-Fernández, M.; Riseley, J.R.; Gillies, G.; Pope, K.; Van Roozendaal, H.; Heng, J.I.; Mandelstam, S.A.; McGillivray, G.; et al. Familial cortical dysplasia caused by mutation in the mammalian target of rapamycin regulator NPRL3. Ann. Neurol. 2016, 79, 132–137. [Google Scholar] [CrossRef]
- Benova, B.; Sanders, M.W.; Uhrova-Meszarosova, A.; Belohlavkova, A.; Hermanovska, B.; Novak, V.; Stanek, D.; Vlckova, M.; Zamecnik, J.; Aronica, E.; et al. GATOR1-related focal cortical dysplasia in epilepsy surgery patients and their families: A possible gradient in severity? Eur. J. Paediatr. Neurol. 2021, 30, 88–96. [Google Scholar] [CrossRef]
- Bennett, M.F.; Hildebrand, M.S.; Kayumi, S.; Corbett, M.A.; Gupta, S.; Ye, Z.; Krivanek, M.; Burgess, R.; Henry, O.J.; Damiano, J.A.; et al. Evidence for a Dual-Pathway, 2-Hit Genetic Model for Focal Cortical Dysplasia and Epilepsy. Neurol. Genet. 2022, 8, e652. [Google Scholar] [CrossRef]
- Chandrasekar, I.; Tourney, A.; Loo, K.; Carmichael, J.; James, K.; Ellsworth, K.A.; Dimmock, D.; Joseph, M. Hemimegalencephaly and intractable seizures associated with the NPRL3 gene variant in a newborn: A case report. Am. J. Med. Genet. A 2021, 185, 2126–2130. [Google Scholar] [CrossRef]
- Tesi, B.; Boileau, C.; Boycott, K.M.; Canaud, G.; Caulfield, M.; Choukair, D.; Hill, S.; Spielmann, M.; Wedell, A.; Wirta, V.; et al. Precision medicine in rare diseases: What is next? J. Intern. Med. 2023, 294, 397–412. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| No. | NPRL3 cDNA Variant and Protein Alteration | Age of Seizure Onset | Sex | Seizure Type | Time to Clinical Diagnosis | Time to Genetic Diagnosis | No. of ASM | Histopathology | Radiological Distribution of FCD | Type of Surgery | Epilepsy Surgery Outcome | Comorbidities |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | NPRL3:c.(?_-21)_(*21_?)del(arr[hg19] 16p13.3(88165_194845)x1) Full gene deletion | 2 years | M | Left temporal plus epilepsy | 5 years | 2 years | 3 | NYD | Left mid-posterior parahippocampal dysplasia | Pending | N/A | Joint hypermobility |
| 2 [20] | c.1375_1376dupAC, p.(S460Pfs*20) Frameshift mutation | 0 months | M | Left temporal epilepsy and bilateral clonic seizures | A few days | N/A | N/A | FCD IIa | Right posterior quadrantic dysplasia | Temporo-parieto-occipital disconnection and frontocentral corticectomy at 11 weeks | Engel II: Partial seizure control at 3 years on oxcarbazepine (residual dysplasia) | Global developmental delay, left hemiplegia, and hemianopia |
| 3 [20] | c.1375_1376dupAC p.(S460Pfs*20) Frameshift mutation | 7 years | F | Right focal lobe epilepsy | N/A | N/A | 2 | FCD IIa | Bottom-of-sulcus dysplasia in the right anterior cingulate sulcus | Resection of FCD at 7 years of age | Engel I: Seizure free at one year after surgery on oxcarbazepine | None |
| 4 [20] | c.1352-4delACAG insTGACCCATCC p.(?) Splicing mutation | 4 months | M | Bilateral tonic and orofacial motor manifestations | N/A | N/A | 3 | FCD IIa | Extensive left frontal operculum and insula dysplasia | Staged resections at 6 and 7 months. Residual dysplasia from the left insula and frontal operculum was resected at 4 years | Engel I: Seizure free and off medication at 6 years | Near normal cognitive and language development and right hemiparesis |
| 5 [20] | c.275G > A p.(R92Q) Missense mutation | 15 months | F | Right frontal lobe epilepsy | N/A | N/A | 2 | FCD IIa | Diffuse dysplasia in the left central head region | Resection was performed at 23 months | Engel I: At 3 years, seizure free and weaning ASM | Mild right hemiparesis and language delay |
| 6 [2] | c.1270C > T p.(R424*) Nonsense mutation | 2 months | NA | Left frontal lobe epilepsy (FFEVF) | 2 months | N/A | 4 | FCD IIa and hippocampal sclerosis | Left frontal lobe FCD | 2 years: postoperative MRI with incomplete resection of FCD, left hippocampal atrophy | Engel II: incomplete FCD resection at 1 year and second surgery at 5 years: rare seizures when medication errors occurred | N/A |
| 7 [2] | c.1070delC p.(P357Hfs*56) Frameshift mutation | 2 years | NA | Right frontal lobe epilepsy (FFEVF) | N/A | N/A | 2 | FCD IIb | Right frontoparietal FCD | No | Engel I: Seizure free at 6 months | N/A |
| 8 [21] | c.973_975del p.(I325del) Inframe deletion | 0 months | F | Left frontal lobe epilepsy | N/A | N/A | 2 | FCD IIa | Left frontal lobe FCD | Resective surgery | Engel I: Seizure free at 1 year 3 months | Moderate ID and impaired motor development |
| 9 [22] | c.48delG p.(S17Afs*70) Frameshift mutation | 2 years | M | Right Frontal lobe epilepsy | 3 years | Post-resection | 1 | FCD IIa | Right postero-mesial frontal FCD | Resection at 6 years of age | Engel III: focal seizures returned at 10 years of age (residual dysplastic) | Inattention and distractibility |
| 10 [22] | c.48delG p.(S17Afs*70) Frameshift mutation | 6 weeks | M | Left frontal lobe epilepsy | 6 weeks | Post-resection | 1 | FCD IIa | Left anteromesial frontal FCD | Focal resection at 2 years | Engel I: seizure free postoperatively, and medication was withdrawn at 3 years | None |
| 11–18 [9] | c.349delG p.(E117Kfs*5) Frameshift mutation | 0 months–15 years | N/A | All had focal onset epilepsy | No single-patient data available | |||||||
| 19 [6] | c.1174C > T p.(Q392*) Nonsense mutation | 10 days of age | F | Epileptic spasms and focal seizures | A few days | 1 year | 7 | N/AN/AN/A | Abnormal signal over left lateral ventricle and a widened left frontotemporal sulcus | Hemispherectomy at 1 year and 2 months of age | Engle I at year | Right-sided cerebral palsy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nabavi Nouri, M.; Alandijani, L.; van Engelen, K.; Tole, S.; Lalonde, E.; Balci, T.B. From Alpha-Thalassemia Trait to NPRL3-Related Epilepsy: A Genomic Diagnostic Odyssey. Genes 2024, 15, 836. https://doi.org/10.3390/genes15070836
Nabavi Nouri M, Alandijani L, van Engelen K, Tole S, Lalonde E, Balci TB. From Alpha-Thalassemia Trait to NPRL3-Related Epilepsy: A Genomic Diagnostic Odyssey. Genes. 2024; 15(7):836. https://doi.org/10.3390/genes15070836
Chicago/Turabian StyleNabavi Nouri, Maryam, Lama Alandijani, Kalene van Engelen, Soumitra Tole, Emilie Lalonde, and Tugce B. Balci. 2024. "From Alpha-Thalassemia Trait to NPRL3-Related Epilepsy: A Genomic Diagnostic Odyssey" Genes 15, no. 7: 836. https://doi.org/10.3390/genes15070836