
The Development of a Fluorescent Microsatellite Marker Assay for the Pitaya Canker Pathogen (Neoscytalidium dimidiatum)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and Isolation
2.2. DNA Extraction
2.3. Isolate Identification
2.4. Genome Assembly and Annotation
2.5. Microsatellite Marker Development and Primer Design
3. Results
3.1. Isolate Identification
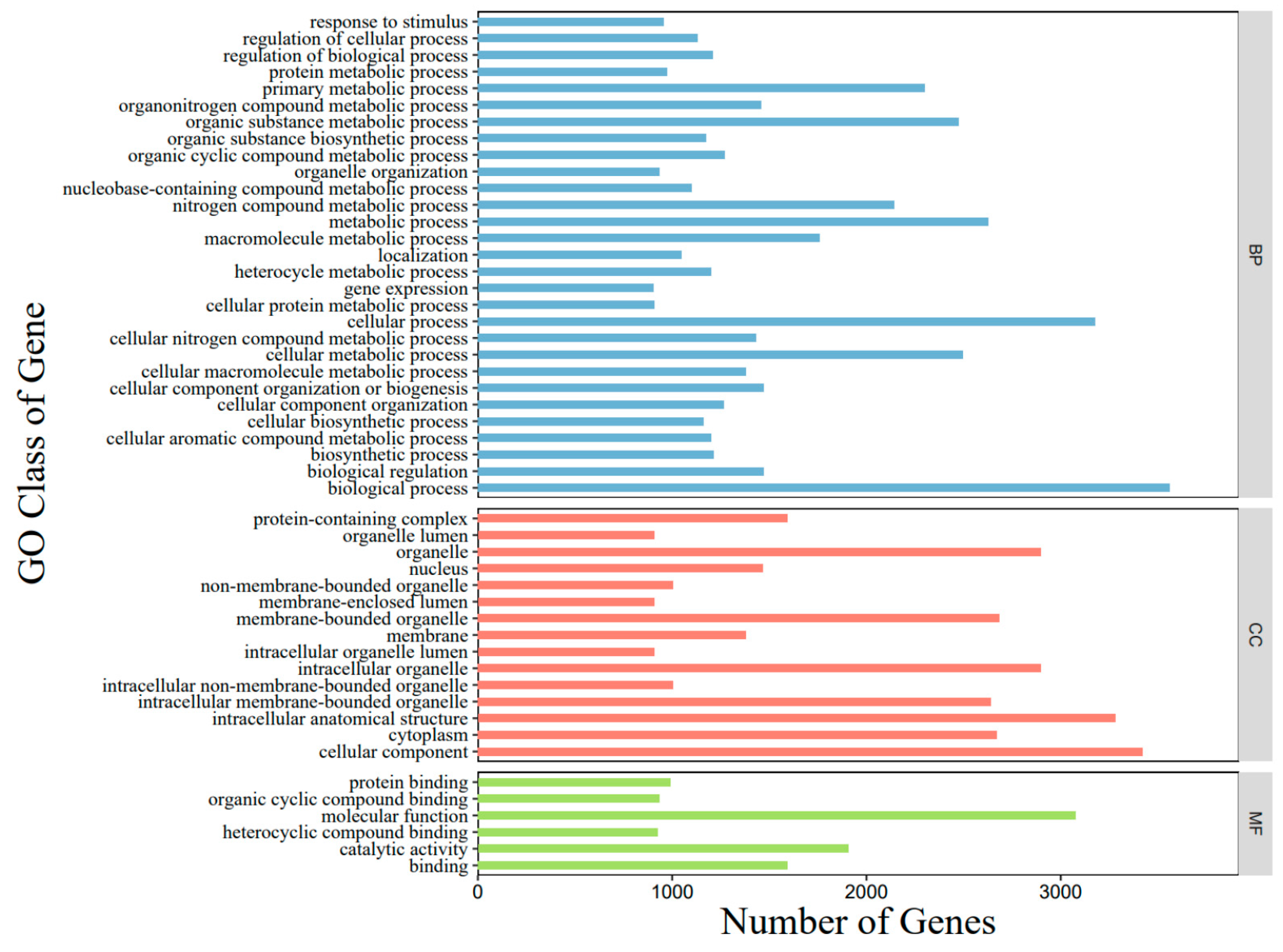
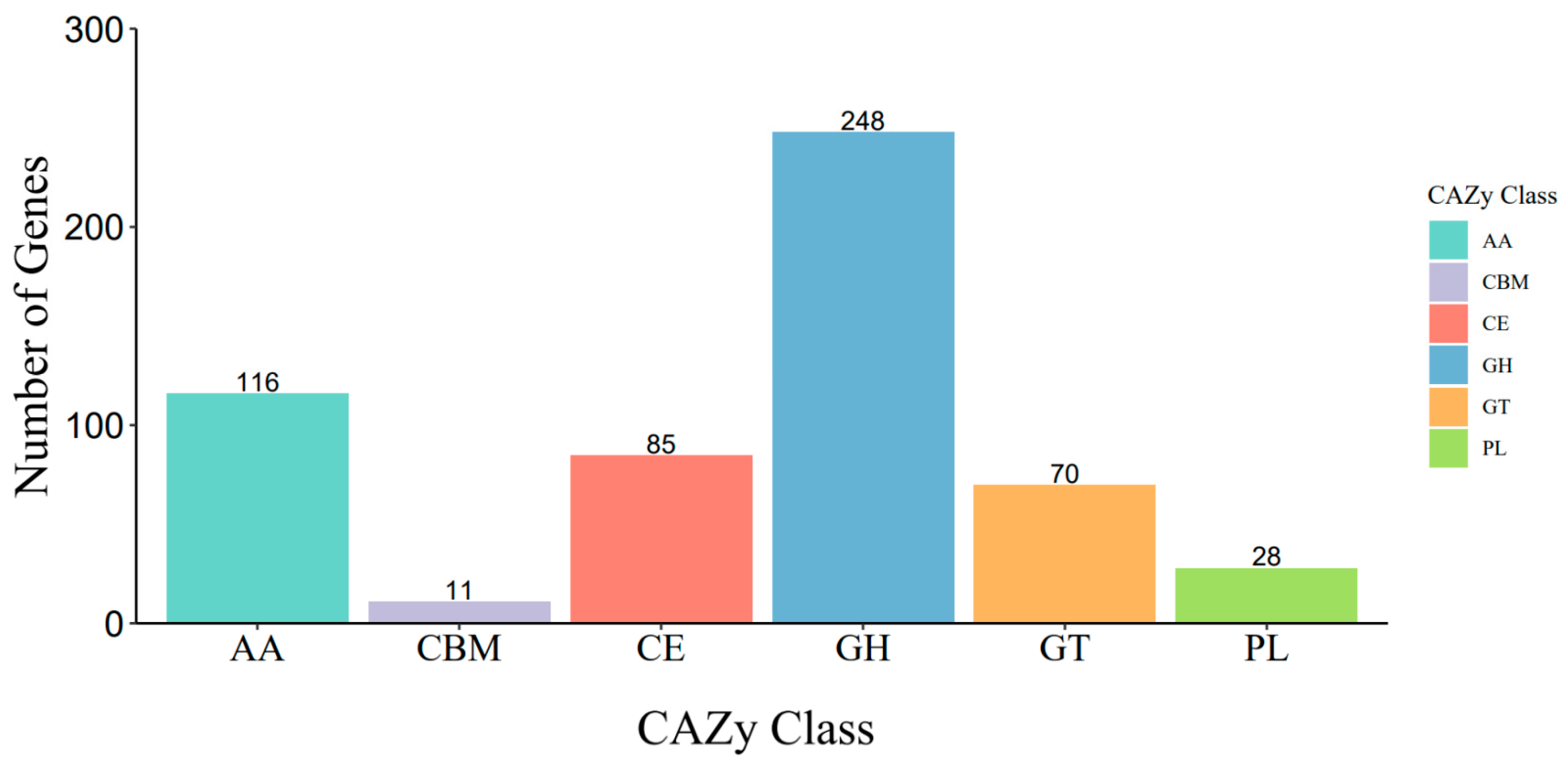
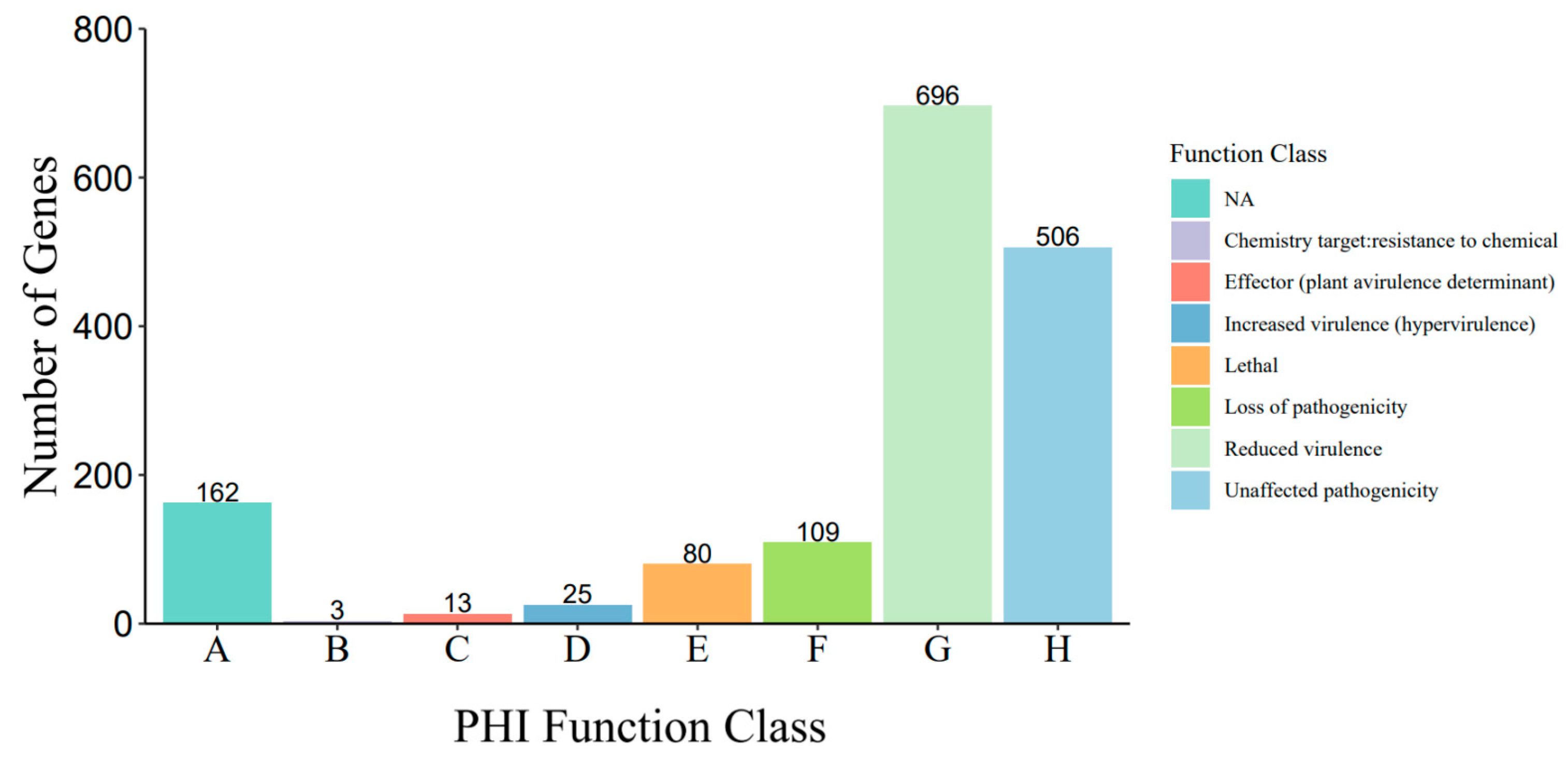
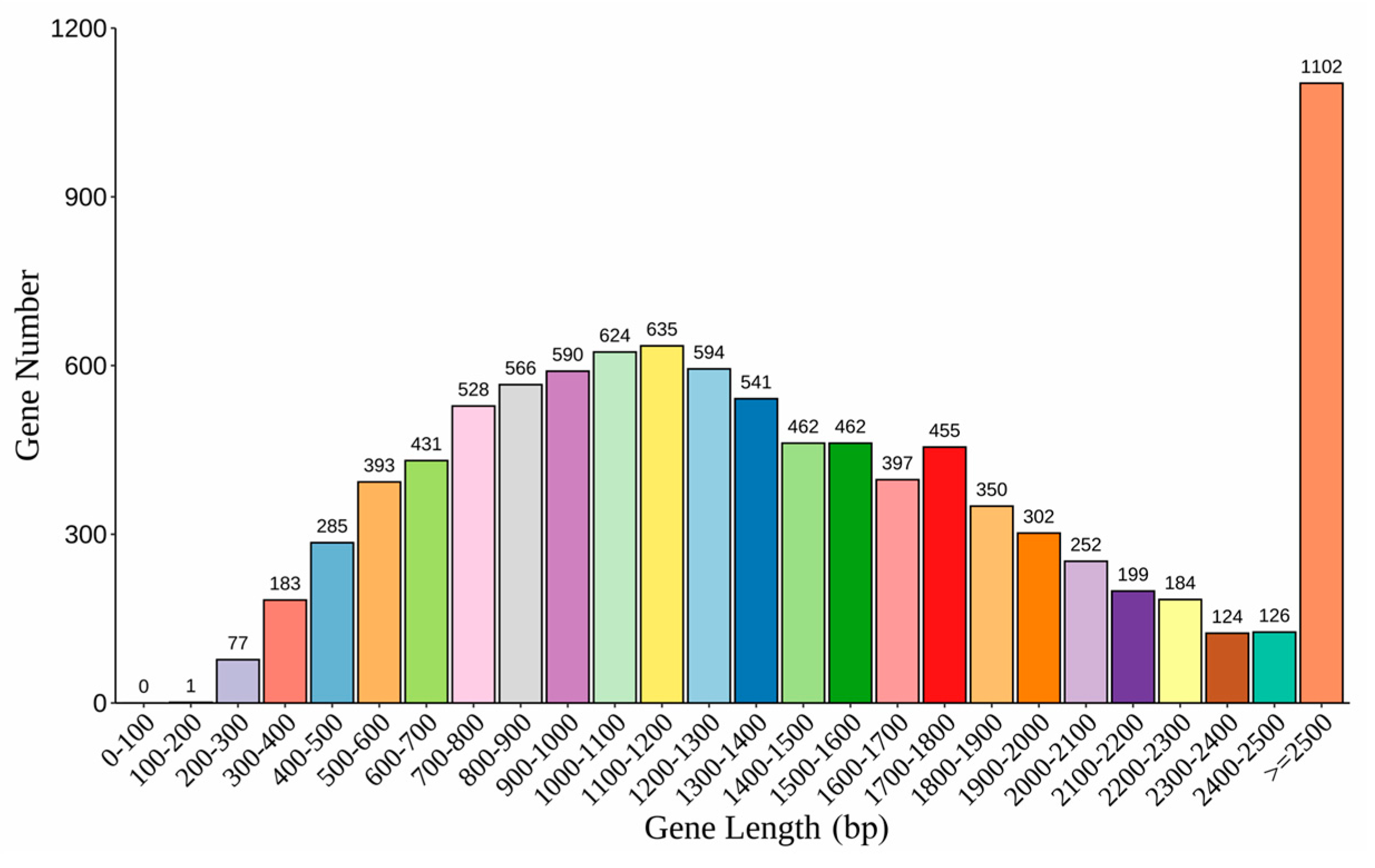
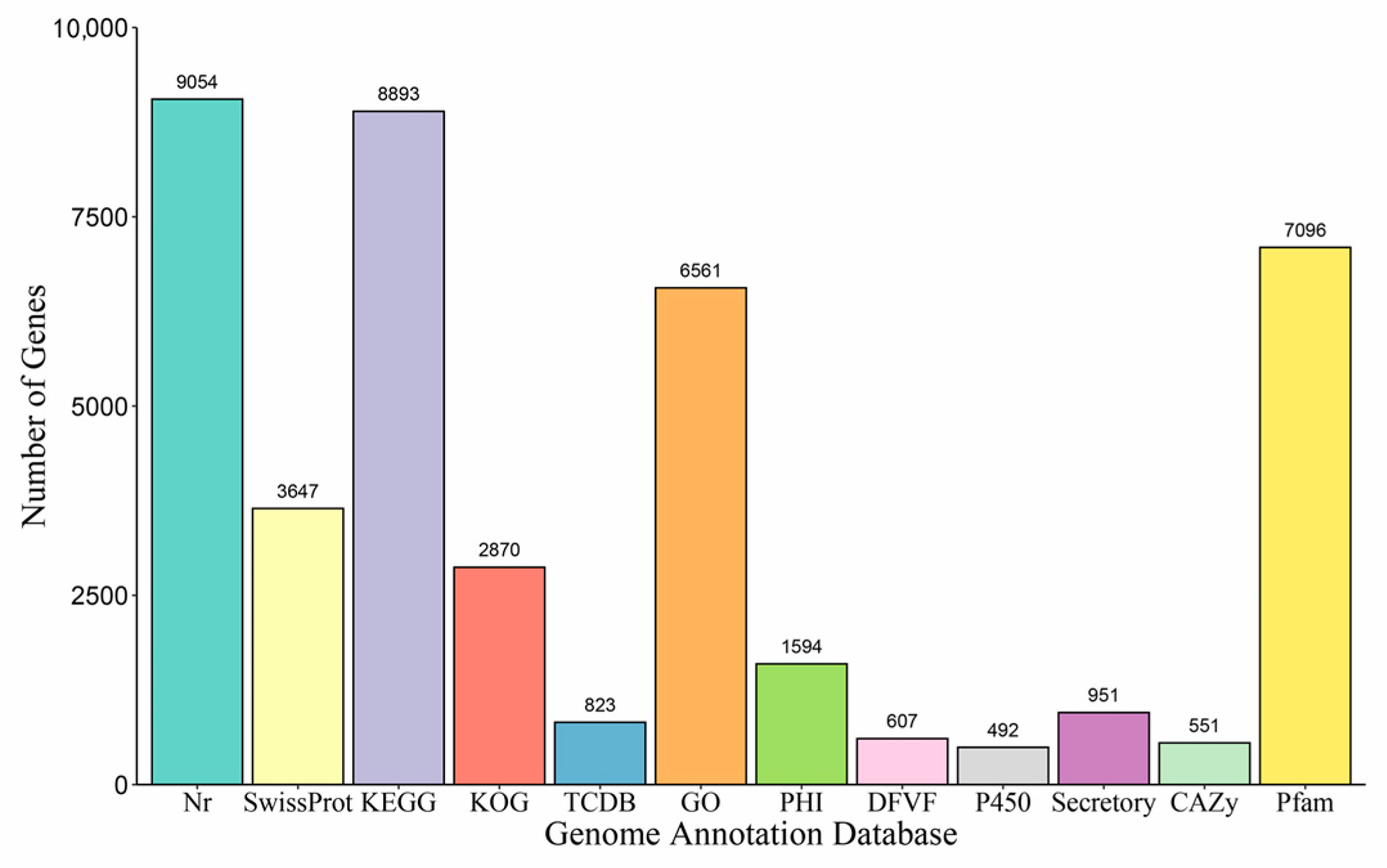
3.2. Genome Assembly and Annotation
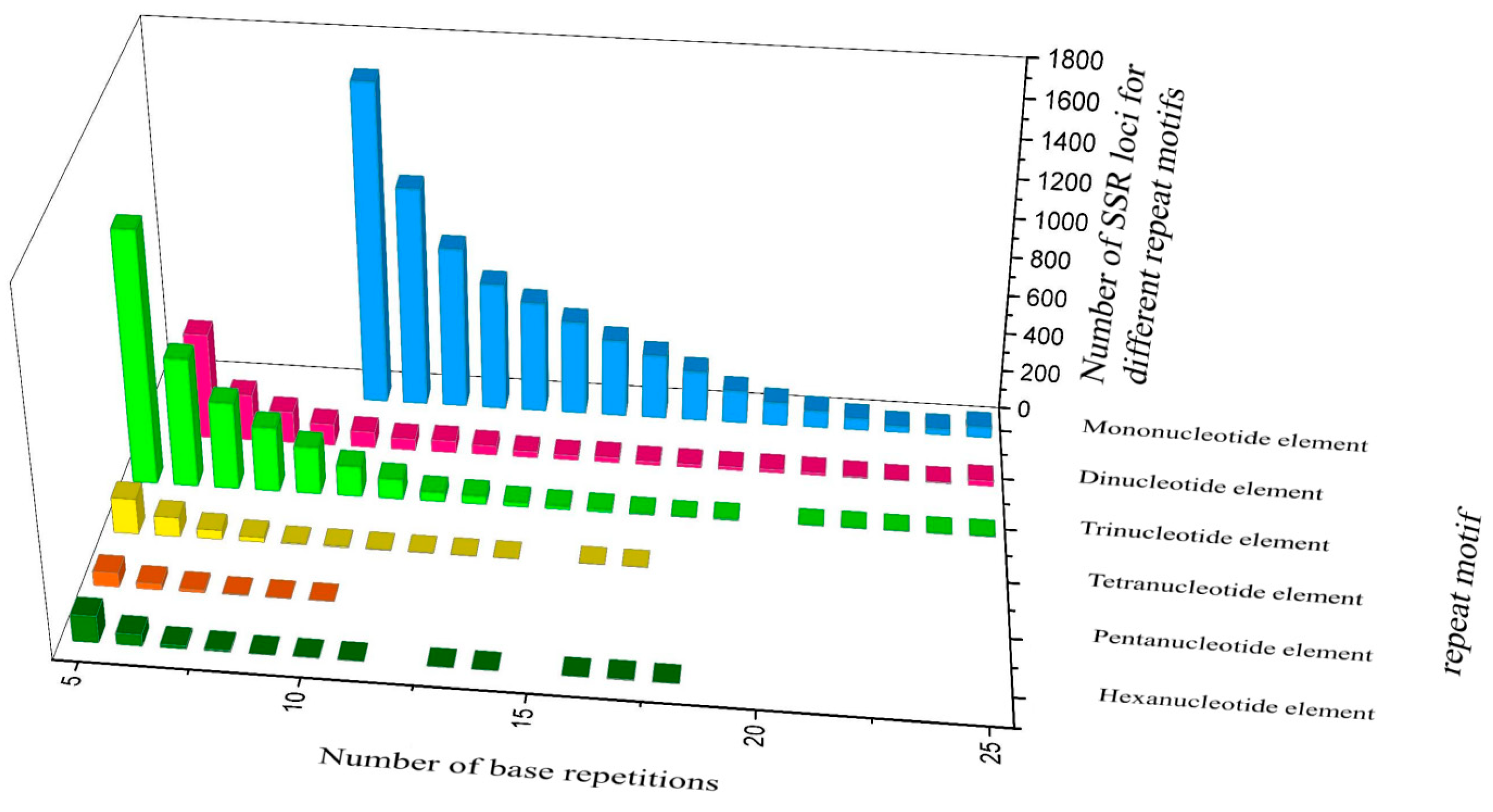
3.3. Polymorphic Microsatellite Markers
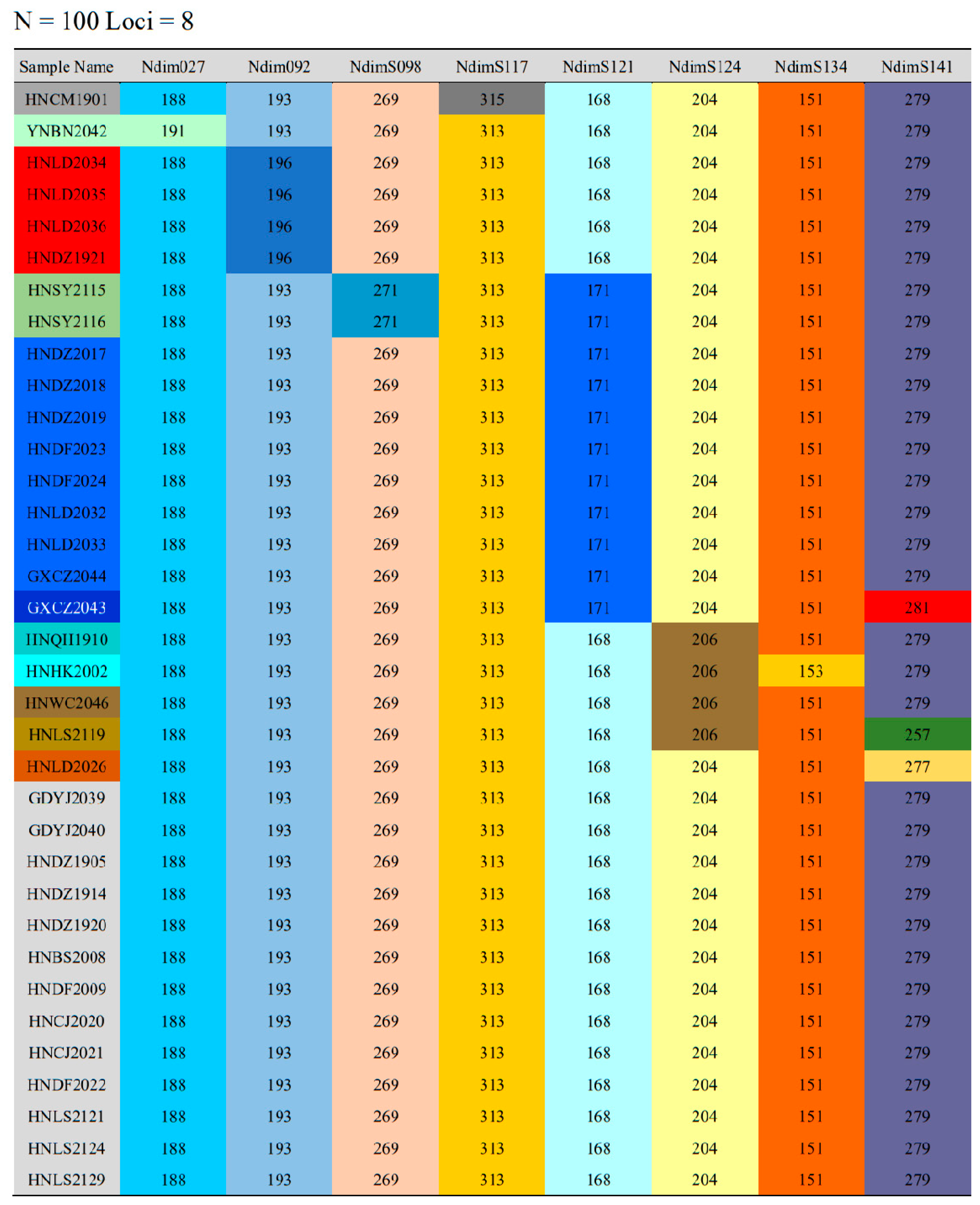
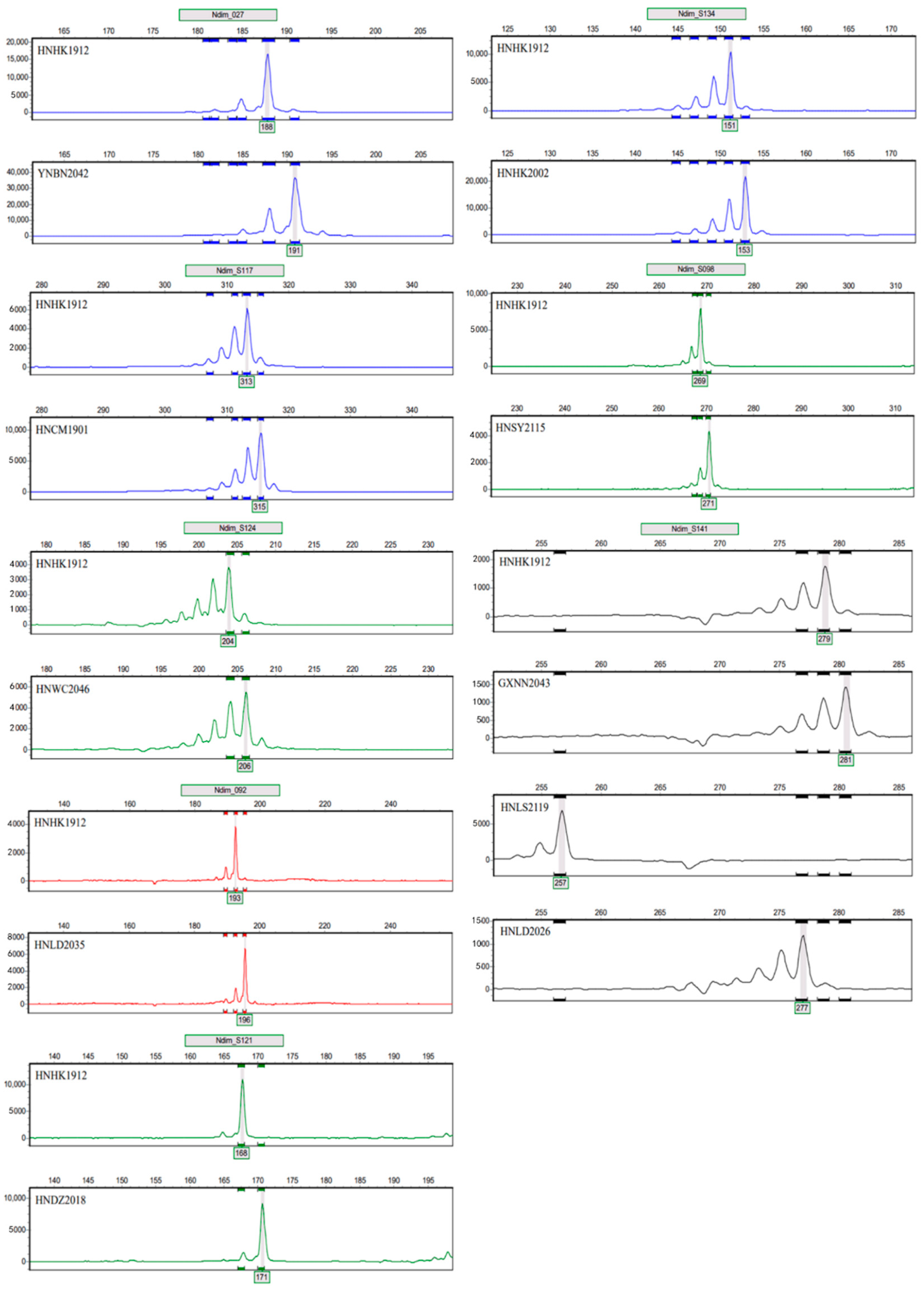
3.4. Validation of the Fluorescent Microsatellite Genotyping Assay
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A





References
- Morton, J.F. (Ed.) Fruits of Warm Climates; Echo Point Books and Media: Brattleboro, VT, USA, 1987; p. 1514. [Google Scholar]
- Le Bellec, F.; Vaillant, F.; Imbert, E. Pitahaya (Hylocereus spp.): A new fruit crop, a market with a future. Fruits 2006, 61, 37–250. [Google Scholar] [CrossRef]
- Adnan, L.; Osman, A.; Abdul, H.A. Antioxidant activity of different extracts of red pitaya (Hylocereus polyrhizus) seed. Int. J. Food Prop. 2011, 14, 1171–1181. [Google Scholar] [CrossRef]
- Ministry of Commerce of the People’s Republic of China. The Tenfold Increase in the Planting Area of Dragon Fruit in China Has Raised Concerns in Vietnam. Available online: http://hochiminh.mofcom.gov.cn/article/jmxw/202012/20201203024850.shtml (accessed on 21 December 2020).
- Produce Report. China’s Dragon Fruit Production Ranks First in the World, with the Fastest Growth Rates in Guangdong, Guangxi, and Hainan Provinces. Available online: https://guojiguoshu.com/article/8128 (accessed on 22 February 2023).
- Yi, R.; Gan, L.; Yan, D.; Wu, Z.; Tong, Y.; Wu, F. Identification and biological characteristics of Neoscytalidium dimidiatum causing pitaya canker. Acta Phytophylacica Sin. 2013, 40, 102–108. [Google Scholar]
- Derviş, S.; Özer, G. Plant-associated Neoscytalidium dimidiatum-taxonomy, host range, epidemiology, virulence, and management strategies: A comprehensive review. J. Fungi 2023, 9, 1048. [Google Scholar] [CrossRef] [PubMed]
- Sanahuja, G.; Lopez, P.; Palmateer, A.J. First report of Neoscytalidium dimidiatum causing stem and fruit canker of Hylocereus undatus in Florida. Plant Dis. 2016, 100, 1499. [Google Scholar] [CrossRef]
- Dai, J.; Wang, H.; Fu, B.; Wang, M.; Tao, T.; Xie, C.; Zhu, C. Determination of biological characteristics of Neoscytalidium dimidiatum and Fusicoccum sp. on pitaya. South China Fruits 2017, 46, 78–82. [Google Scholar]
- Zhang, R.; Liu, A.; Bai, C.; Liu, C.; Luo, X.; Jiang, F.; Liang, R.; Zheng, H. Symptom observation and pathogen identification on canker disease of pitaya. J. Fruit Sci. 2013, 30, 854–856. [Google Scholar]
- Zhu, W.; Zhan, J. Population genetics of plant pathogens. Hereditas 2012, 34, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Marwah, V.S.; Yadav, A.; Jha, V.; Dhaygude, K.; Bangar, U.; Kulkarni, V.; Jere, A. Identification of optimum sequencing depth especially for de novo genome assembly of small genomes using next generation sequencing data. PLoS ONE 2013, 8, e60204. [Google Scholar] [CrossRef]
- Litt, M.; Luty, J.A. A hypervariable microsatellite revealed by in vitro amplification of a dinucleotide repeat within the cardiac muscle actin gene. Am. J. Hum. Genet. 1989, 44, 397–401. [Google Scholar]
- Fan, C.; Liu, Q.; Zeng, B.; Qiu, Z.; Zhou, C.; Chen, K.; Guo, G. Development of simple sequence repeat (SSR) markers and genetic diversity analysis in blackwood clones in China. Silvae Genet. 2016, 65, 49–54. [Google Scholar] [CrossRef]
- Tsykun, T.; Rellstab, C.; Dutech, C.; Sipos, G.; Prospero, S. Comparative assessment of SSR and SNP markers for inferring the population genetic structure of the common fungus Armillaria cepistipes. Heredity 2017, 119, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Guichoux, E.; Lagache, L.; Wagner, S.; Chaumeil, P.; Léger, P.; Lepais, O.; Lepoittevin, C.; Malausa, T.; Revardel, E.; Salin, F.; et al. Current trends in microsatellite genotyping. Mol. Ecol. Resour. 2011, 11, 591–611. [Google Scholar] [CrossRef]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Salehi, M.; Arzani, A.; Talebi, M.; Rokhzadi, A. Genetic diversity of wheat wild relatives using SSR markers. Genetika 2018, 50, 131–141. [Google Scholar] [CrossRef]
- Prasanna, H.C.; Sinha, D.P.; Verma, A.; Singh, M.; Singh, B.; Rai, M.; Martin, D.P. The population genomics of begomoviruses: Global scale population structure and gene flow. Virol. J. 2010, 7, 220–232. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S. Analysis of phylogenetic relationships by amplification and direct sequencing of ribosomal RNA genes. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; pp. 315–322. [Google Scholar]
- Glass, N.L.; Donaldson, G.C. Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl. Environ. Microbiol. 1995, 61, 1323–1330. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Gao, F.; Jakovlić, I.; Lei, H.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G.; Zhang, D. Using PhyloSuite for molecular phylogeny and tree-based analyses. iMeta 2023, 2, e87. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Xu, H.; Luo, X.; Qian, J.; Pang, X.; Song, J.; Qian, G.; Chen, J.; Chen, S. FastUniq: A fast de novo duplicates removal tool for paired short reads. PLoS ONE 2012, 7, e52249. [Google Scholar] [CrossRef]
- Liu, Y.; Schröder, J.; Schmidt, B. Musket: A multistage k-mer spectrum-based error corrector for Illumina sequence data. Bioinformatics 2013, 29, 308–315. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Liu, B.; Shi, Y.; Yuan, J.; Hu, X.; Zhang, H.; Li, N.; Fan, W. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv 2013, arXiv:1308.2012. [Google Scholar]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Thomas, C.M. Improved Genome Assembly and annotation of the soybean aphid (Aphis glycines Matsumura). G3-Genes Genomes Genet. 2020, 10, 899–906. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Bridges, S.; Magbanua, Z.V.; Peterson, D.G. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 2008, 36, 2284–2294. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Lu, Z.; Wang, S.; Wang, J.J.-Y.; Gao, X. CMsearch: Simultaneous exploration of protein sequence space and structure space improves not only protein homology detection but also protein structure prediction. Bioinformatics 2016, 32, i332–i340. [Google Scholar] [CrossRef] [PubMed]
- Burge, S.W.; Daub, J.; Eberhardt, R.; Tate, J.; Barquist, L.; Nawrocki, E.P.; Eddy, S.R.; Gardner, P.P.; Bateman, A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013, 41, D226–D232. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinformatics 2009, 10, 421. [Google Scholar] [CrossRef]
- Li, W.; Jaroszewski, L.; Godzik, A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 2002, 18, 77–82. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinformatics 2003, 4, 41. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.; Eddy, S.R.; Durbin, R. Pfam: A comprehensive database of protein domain families based on seed alignments. Proteins 1997, 28, 405–420. [Google Scholar] [CrossRef]
- Saier, M.H.; Reddy, V.S.; Moreno-Hagelsieb, G.; Hendargo, K.J.; Zhang, Y.; Iddamsetty, V.; Lam, K.J.K.; Tian, N.; Russum, S.; Wang, J.; et al. The transporter classification database (TCDB): 2021 update. Nucleic Acids Res. 2021, 49, D461–D467. [Google Scholar] [CrossRef]
- Lisitsa, A.V.; Gusev, S.A.; Karuzina, I.I.; Archakov, A.I.; Koymans, L. Cytochrome P450 database. SAR QSAR Environ. Res. 2001, 12, 359–366. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; De Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Urban, M.; Pant, R.; Raghunath, A.; Irvine, A.G.; Pedro, H.; Hammond-Kosack, K.E. The pathogen-host interactions database (PHI-base): Additions and future developments. Nucleic Acids Res. 2015, 43, D645–D655. [Google Scholar] [CrossRef]
- Lu, T.; Yao, B.; Zhang, C. DFVF: Database of fungal virulence factors. Database 2012, 2012, bas032. [Google Scholar] [CrossRef]
- Jiang, S.; Luo, J.; Wang, X.; Shi, C. A study on efficient screening of the primers for selecting polymorphic SSR markers based on the re-sequencing data in Pyrus. J. Fruit Sci. 2019, 36, 129–136. [Google Scholar]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Paisley, B.M.; Liu, Y. GeneMarkeR: A database and user interface for scRNA-seq marker genes. Front. Genet. 2021, 12, 763431. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. Peer-J 2014, 2, e281. [Google Scholar] [CrossRef]
- Wonglom, P.; Pornsuriya, C.; Sunpapao, A. A new species of Neoscytalidium hylocereum sp. nov. causing canker on red-fleshed dragon fruit (Hylocereus polyrhizus) in Southern Thailand. J. Fungi 2023, 9, 197. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, G.; Lin, F.; Wang, Z.; Jin, G.; Yang, L.; Wang, Y.; Chen, X.; Xu, Z.; Zhao, X.; et al. Development of microsatellite markers and construction of genetic map in rice blast pathogen Magnaporthe grisea. Fungal Genet. Biol. 2008, 45, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gao, S.; Liu, Y.; Wang, P.; Zhang, Z.; Chen, D. A pipeline for effectively developing highly polymorphic simple sequence repeats markers based on multi-sample genomic data. Ecol. Evol. 2022, 12, e8705. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Wang, X.; Zhan, G.; Wei, G.; Zhou, X.; Zhao, J.; Huang, L.; Kang, Z. Genome-Wide analysis of simple sequence sepeats and efficient development of polymorphic SSR markers based on whole genome re-sequencing of multiple isolates of the wheat stripe rust fungus. PLoS ONE 2015, 10, e0130362. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolates | Years | Locations | Reference Genome | Resequencing | Primer Screening |
|---|---|---|---|---|---|
| HNCM1901 | 2019 | Chengmai, Hainan | - | - | Yes |
| HNDZ1905 | 2019 | Danzhou, Hainan | - | - | - |
| HNQH1910 | 2019 | Qionghai, Hainan | - | - | Yes |
| HNDZ1914 | 2019 | Danzhou, Hainan | - | - | Yes |
| HNDZ1920 | 2019 | Danzhou, Hainan | Yes | - | - |
| HNDZ1921 | 2019 | Danzhou, Hainan | - | - | - |
| HNHK2002 | 2020 | Haikou, Hainan | - | - | Yes |
| HNBS2008 | 2020 | Baisha, Hainan | - | - | Yes |
| HNDF2009 | 2020 | Dongfang, Hainan | - | - | - |
| HNDZ2017 | 2020 | Danzhou, Hainan | - | - | - |
| HNDZ2018 | 2020 | Danzhou, Hainan | - | - | - |
| HNDZ2019 | 2020 | Danzhou, Hainan | - | - | - |
| HNCJ2020 | 2020 | Changjiang, Hainan | - | - | Yes |
| HNCJ2021 | 2020 | Changjiang, Hainan | - | - | - |
| HNDF2022 | 2020 | Dongfang, Hainan | - | - | Yes |
| HNDF2023 | 2020 | Dongfang, Hainan | - | - | - |
| HNDF2024 | 2020 | Dongfang, Hainan | - | - | - |
| HNLD2026 | 2020 | Ledong, Hainan | - | - | - |
| HNLD2032 | 2020 | Ledong, Hainan | - | - | - |
| HNLD2033 | 2020 | Ledong, Hainan | - | - | - |
| HNLD2034 | 2020 | Ledong, Hainan | - | Yes | - |
| HNLD2035 | 2020 | Ledong, Hainan | - | Yes | - |
| HNLD2036 | 2020 | Ledong, Hainan | - | Yes | - |
| GDYJ2039 | 2020 | Yangjiang, Hainan | - | Yes | Yes |
| GDYJ2040 | 2020 | Yangjiang, Hainan | - | - | - |
| YNBN2042 | 2020 | Xishuangbanna, Yunnan | - | - | Yes |
| GXCZ2043 | 2020 | Chongzuo, Guangxi | - | - | Yes |
| GXCZ2044 | 2020 | Chongzuo, Guangxi | - | - | - |
| HNWC2046 | 2020 | Wenchang, Hainan | - | - | Yes |
| HNSY2115 | 2021 | Sanya, Hainan | - | - | - |
| HNSY2116 | 2021 | Sanya, Hainan | - | Yes | Yes |
| HNLS2119 | 2021 | Lingshui, Hainan | - | - | - |
| HNLS2121 | 2021 | Lingshui, Hainan | - | - | - |
| HNLS2124 | 2021 | Lingshui, Hainan | - | - | Yes |
| HNLS2129 | 2021 | Lingshui, Hainan | - | - | - |
| Species | Voucher/Culture | Location | GenBank Accession Number | |
|---|---|---|---|---|
| ITS | tub | |||
| N. dimidiatum | Arp2-D | Turkey | MK813852 | MK813852 |
| N. dimidiatum | Kale4-C | Turkey | MK788362 | MK788362 |
| N. dimidiatum | Nd_Fig01 | Turkey | OL304243 | OK788660 |
| N. dimidiatum | Nd_Fig02 | Turkey | OL304244 | OK788661 |
| N. dimidiatum | Nd_Fig03 | Turkey | OL304245 | OK788662 |
| N. dimidiatum | Ol_Dr04 | Turkey | OK416080 | OK428827 |
| N. dimidiatum | Ol_Hl02 | Turkey | OK416072 | OK428819 |
| N. dimidiatum | Ol_Nz19 | Turkey | OK416079 | OK428826 |
| N. dimidiatum | Nd_Pp01 | Turkey | OK643641 | OK666382 |
| N. hyalinum | CBS 125608 | Gabon | MH863571 | MT592752 |
| N. hyalinum | CBS 125609 | Gabon | MH863572 | MT592753 |
| N. hyalinum | CBS 125610 | Gabon | MH863573 | MT592754 |
| N. hyalinum | CBS 125617 | France | MH863577 | MT592756 |
| Neoscytalidium sp. | CBS 125619 | France | MT587533 | MT592757 |
| Neoscytalidium sp. | CBS 125622 | Martinique | MT587537 | MT592765 |
| N. hyalinum | CBS 125623 | Martinique | MH863579 | MT592766 |
| N. dimidiatum | CBS 125695 | France | KX464231 | KX465065 |
| N. hyalinum | CBS 125808 | Martinique | MH863768 | MT592767 |
| Neoscytalidium sp. | CBS 137.77 | USA | MT587535 | MT592763 |
| S. hyalinum | CBS 145.78 | UK | KF531816 | KF531796 |
| N. dimidiatum | CBS 251.49 | USA | KF531819 | KF531799 |
| Neoscytalidium sp. | CBS 312.90 | Netherlands | MT587536 | MT592764 |
| N. dimidiatum | CBS 499.66 | Mali | KF531820 | KF531800 |
| Neoscytalidium sp. | CBS 601.85 | USA | MT587538 | MT592768 |
| Neoscytalidium sp. | CBS 662.77 | - | MT587534 | MT592758 |
| N. hyalinum | CMM 3566 | Brazil | KF234551 | KF254935 |
| N. hyalinum | CMM 3649 | Brazil | KF234550 | KF254934 |
| N. hyalinum | COUFAL 0144 | Brazil | MH251953 | MH251969 |
| N. hyalinum | COUFAL 0145 | Brazil | MH251954 | MH251970 |
| N. hyalinum | COUFAL 0146 | Brazil | MH251955 | MH251971 |
| N. dimidiatum | DE 1606 | China | KY013660 | KY349087 |
| N. hylocereum | PSU-HP01 | Thailand | LC590859 | LC647832 |
| N. hylocereum | TSU-HP01 | Thailand | LC590860 | LC647833 |
| N. hylocereum | TSU-HP02 | Thailand | LC590861 | LC647834 |
| N. novaehollandiae | CBS 122070 | Australia | EF585539 | MT592759 |
| N. novaehollandiae | CBS 122071 | Australia | EF585540 | MT592760 |
| N. novaehollandiae | CBS 122072 | Australia | EF585535 | MT592761 |
| N. novaehollandiae | CBS 122610 | Australia | EF585536 | MT592762 |
| Neoscytalidium sp. | IOM 325287 | Mexico | MG764431 | - |
| N. orchidacearum | MFLUCC 12-0533 | Thailand | KU179865 | - |
| N. dimidiatum | HNDZ1920 | China | PP917774 | PP928423 |
| N. dimidiatum | HNLD2034 | China | PP917775 | PP928424 |
| N. dimidiatum | HNLD2035 | China | PP917776 | PP928425 |
| N. dimidiatum | HNLD2036 | China | PP917777 | PP928426 |
| N. dimidiatum | GDYJ2039 | China | PP917778 | PP928427 |
| N. dimidiatum | HNSY2116 | China | PP917779 | PP928428 |
| B. dothidea (Outgroup) | CMW 8000 | Switzerland | AY236949 | AY236927 |
| Genome Features | N. dimidiatum Genome |
|---|---|
| Number of contigs | 578 |
| Assembled genome size (bp) | 43,460,451 |
| Contig N50 (bp) | 266,892 |
| Contig L50 | 45 |
| Largest contig | 1,170,203 |
| GC content of the genome (%) | 53.96 |
| Ns per 100 kbp | 3.41 |
| Content of repeat sequences (%) | 2,801,256 bp (6.45%) |
| tRNA | 138 |
| rRNA | 15 |
| sRNA | 3 |
| snRNA | 38 |
| Predicted protein-coding genes (#) | 9863 |
| Locus | Primer | Product Length (bp) | Motif | Fluorophore |
|---|---|---|---|---|
| Ndim027 | F:5′-GAGCAAAGGACACCAAAGCG-3′ | 188–191 | AAG | FAM |
| R:5′-GTCTCGATCTTGGTCGTCGG-3′ | ||||
| Ndim092 | F:5′-TCGCACAACACTTCGCAAAG-3′ | 193–196 | AAG | ROX |
| R:5′-TGGATCGACGCCTTTGGAAA-3′ | ||||
| NdimS098 | F:5′-TTGGGTCCAGCTTGTGTTGT-3′ | 269–271 | GA | HEX |
| R:5′-GGTGCCTGCTCATTACGGTA-3′ | ||||
| NdimS117 | F:5′-CTCTTTGTCCGCTGGATGGT-3′ | 313–315 | CT | FAM |
| R:5′-ACAAGCCCCATACCCGTAAC-3′ | ||||
| NdimS121 | F:5′-CATCGAACGCATGCAAGAGG-3′ | 168–171 | ACC | HEX |
| R:5′-GGAGAAAGGCGTGCTCATGT-3′ | ||||
| NdimS124 | F:5′-ACACCTTTCTAGCGCAGTCC-3′ | 204–206 | AG | HEX |
| R:5′-TGAAGGTCTGGTCGATGTGC-3′ | ||||
| NdimS134 | F:5′-ATGTCGGCGCGTTATCTGAT-3′ | 151–153 | GT | FAM |
| R:5′-GGGTCCAGAATTCTCACCGG-3′ | ||||
| NdimS141 | F:5′-TCAAACGCTTCCCCTTCCTC-3′ | 257–281 | CT | TAMRA |
| R:5′-TGAGGAAGGAATCGATCGCG-3′ |
| Locus | k | N | PIC |
|---|---|---|---|
| Ndim027 | 2 | 35 | 0.054 |
| Ndim092 | 2 | 35 | 0.182 |
| NdimS098 | 2 | 35 | 0.102 |
| NdimS117 | 2 | 35 | 0.054 |
| NdimS121 | 2 | 35 | 0.338 |
| NdimS124 | 2 | 35 | 0.182 |
| NdimS134 | 2 | 35 | 0.054 |
| NdimS141 | 4 | 35 | 0.158 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Li, X.; Tang, J.; Xie, C.; Wang, J. The Development of a Fluorescent Microsatellite Marker Assay for the Pitaya Canker Pathogen (Neoscytalidium dimidiatum). Genes 2024, 15, 885. https://doi.org/10.3390/genes15070885
Li R, Li X, Tang J, Xie C, Wang J. The Development of a Fluorescent Microsatellite Marker Assay for the Pitaya Canker Pathogen (Neoscytalidium dimidiatum). Genes. 2024; 15(7):885. https://doi.org/10.3390/genes15070885
Chicago/Turabian StyleLi, Rui, Xi Li, Jingcheng Tang, Changping Xie, and Jianan Wang. 2024. "The Development of a Fluorescent Microsatellite Marker Assay for the Pitaya Canker Pathogen (Neoscytalidium dimidiatum)" Genes 15, no. 7: 885. https://doi.org/10.3390/genes15070885
APA StyleLi, R., Li, X., Tang, J., Xie, C., & Wang, J. (2024). The Development of a Fluorescent Microsatellite Marker Assay for the Pitaya Canker Pathogen (Neoscytalidium dimidiatum). Genes, 15(7), 885. https://doi.org/10.3390/genes15070885