
Complete Chloroplast Genome of Megacarpaea megalocarpa and Comparative Analysis with Related Species from Brassicaceae

Abstract
:1. Introduction
2. Materials and Methods
2.1. Survey Site Sampling and DNA Sequencing
2.2. Chloroplast Genome Assembly and Annotation Analyses
2.3. Analysis of SSRs and Repetitive Sequences
2.4. Inverted Repeats Boundary Analysis, Codon Usage Bias, and Genome Comparison
2.5. Ka/Ks Analysis and Nucleotide Diversity
2.6. Phylogenetic Analysis and Estimation of Divergence Times
3. Results
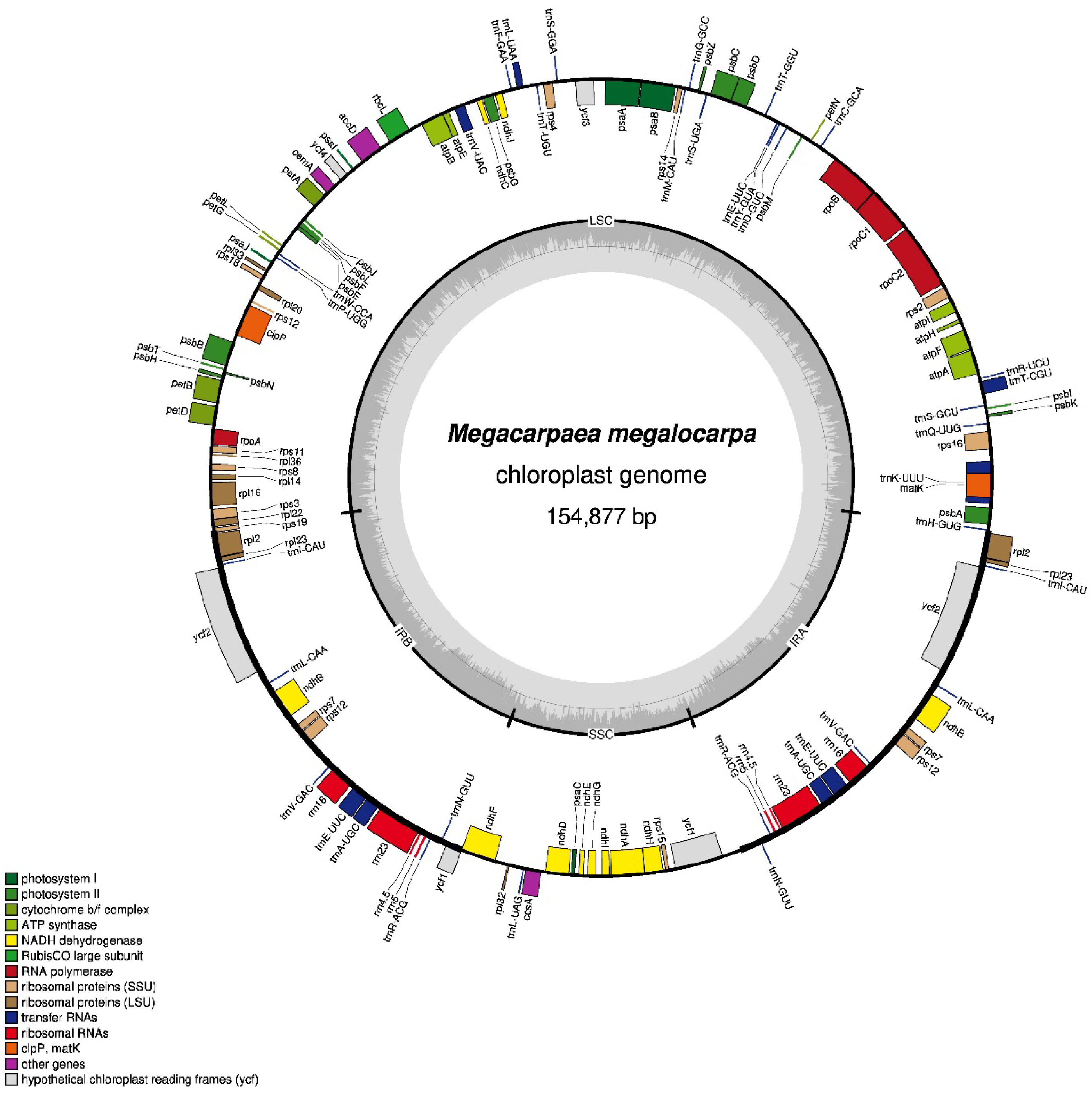
3.1. Basic Characteristics of the M. megalocarpa Chloroplast Genome
3.2. SSRs and Long Repeat Sequences
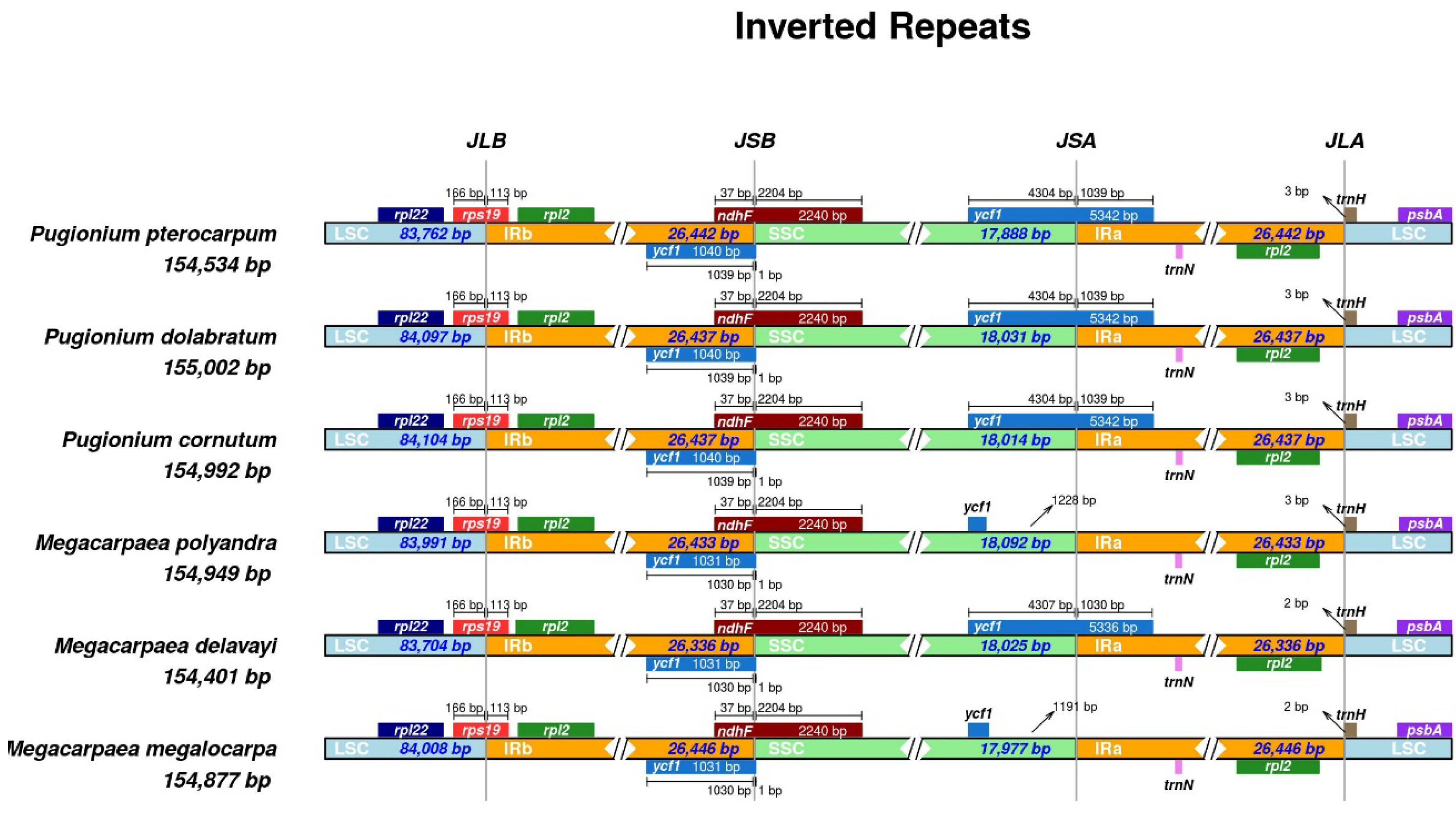
3.3. Expansion and Contraction of the Inverted Repeat Boundaries
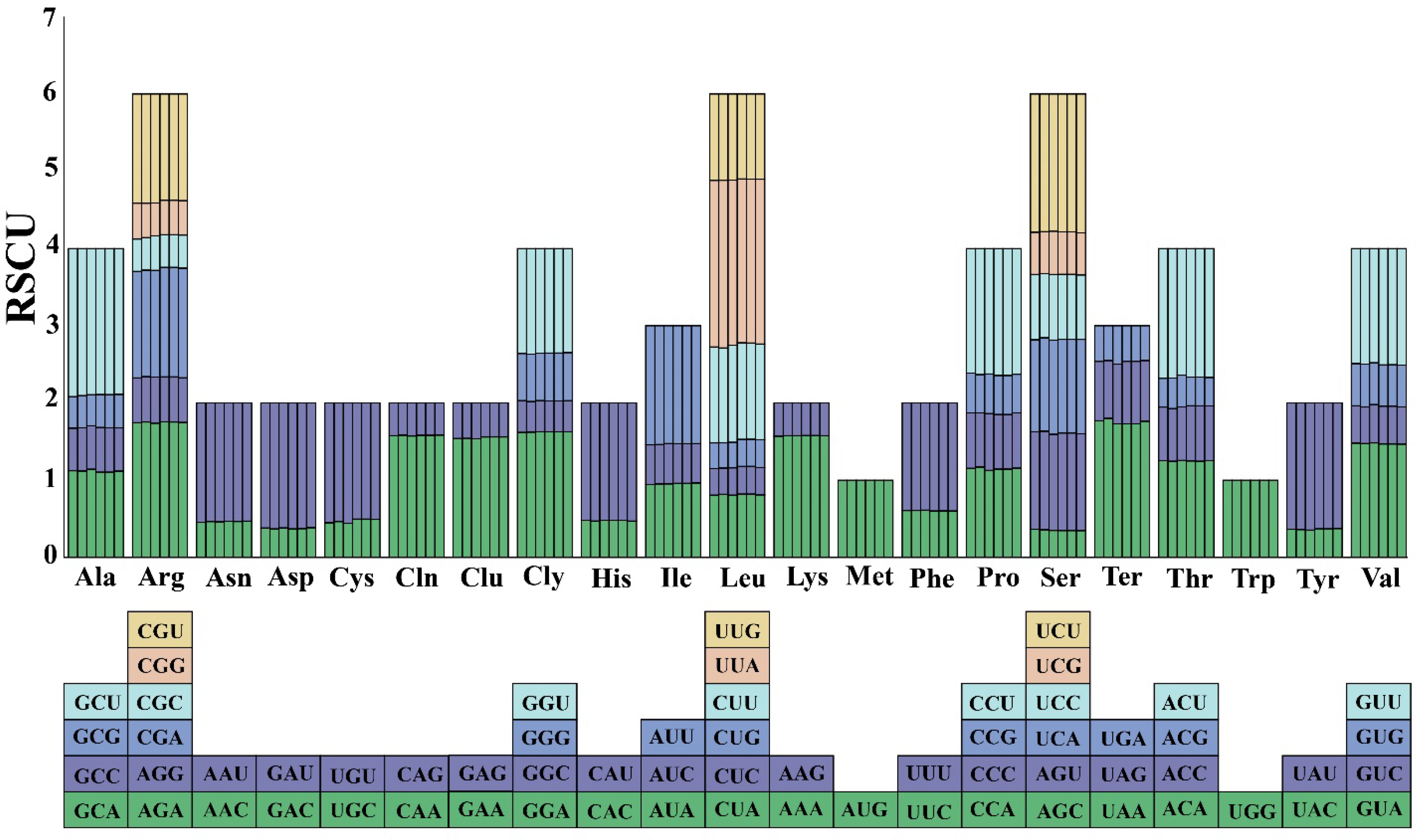
3.4. Codon Usage Bias Analysis
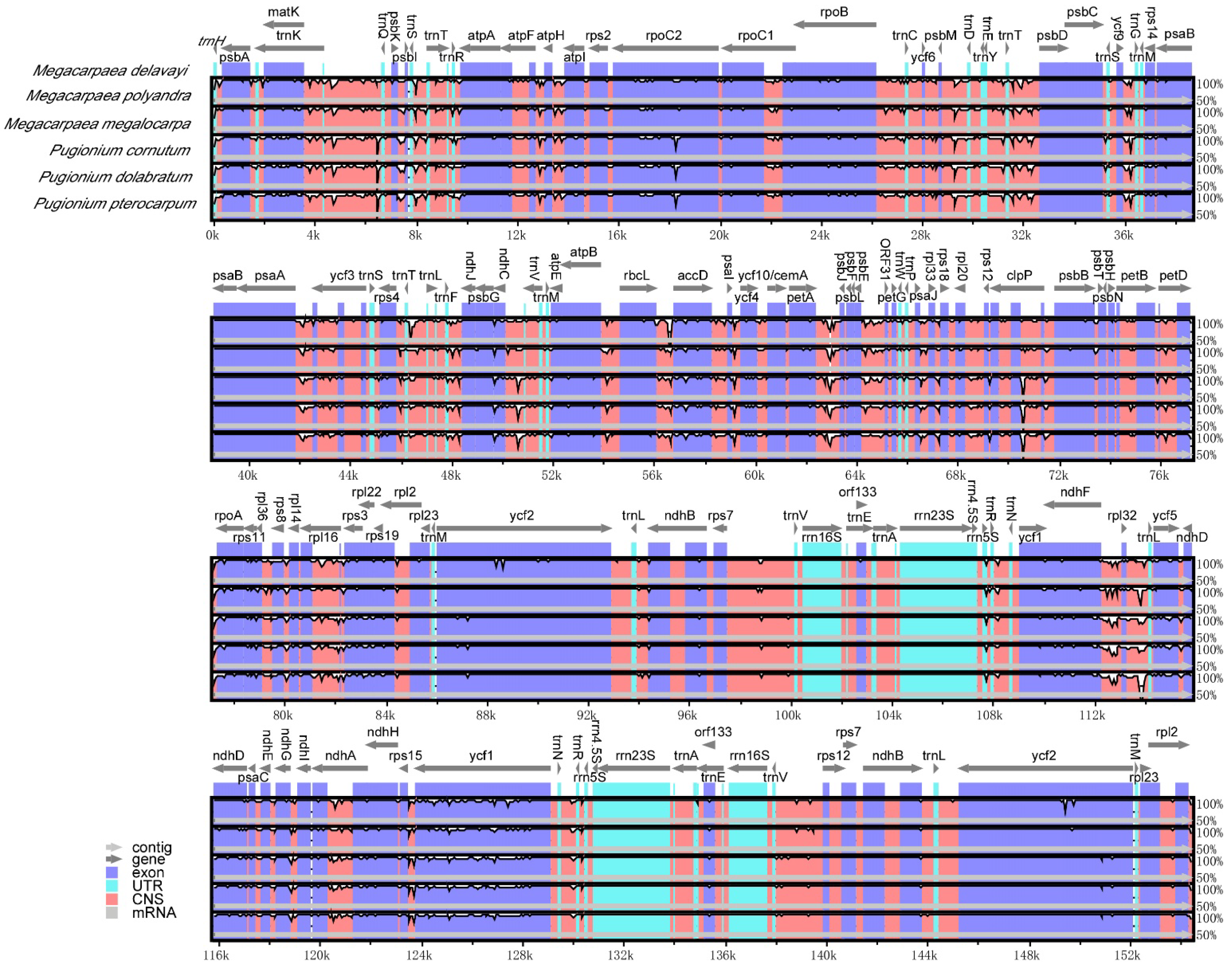
3.5. Comparative Analysis of Chloroplast Genome Sequences
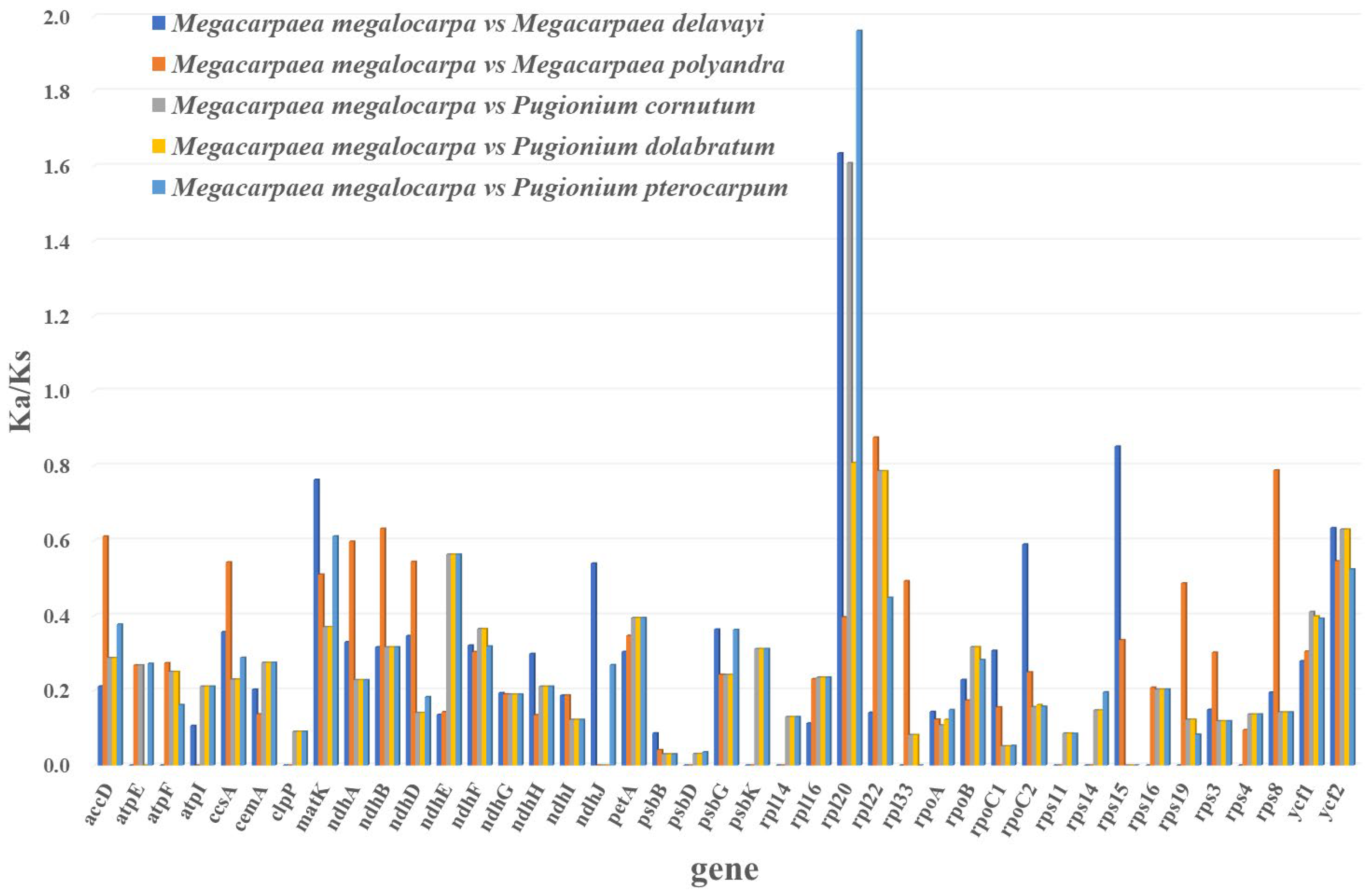
3.6. Selective Pressure Analyses
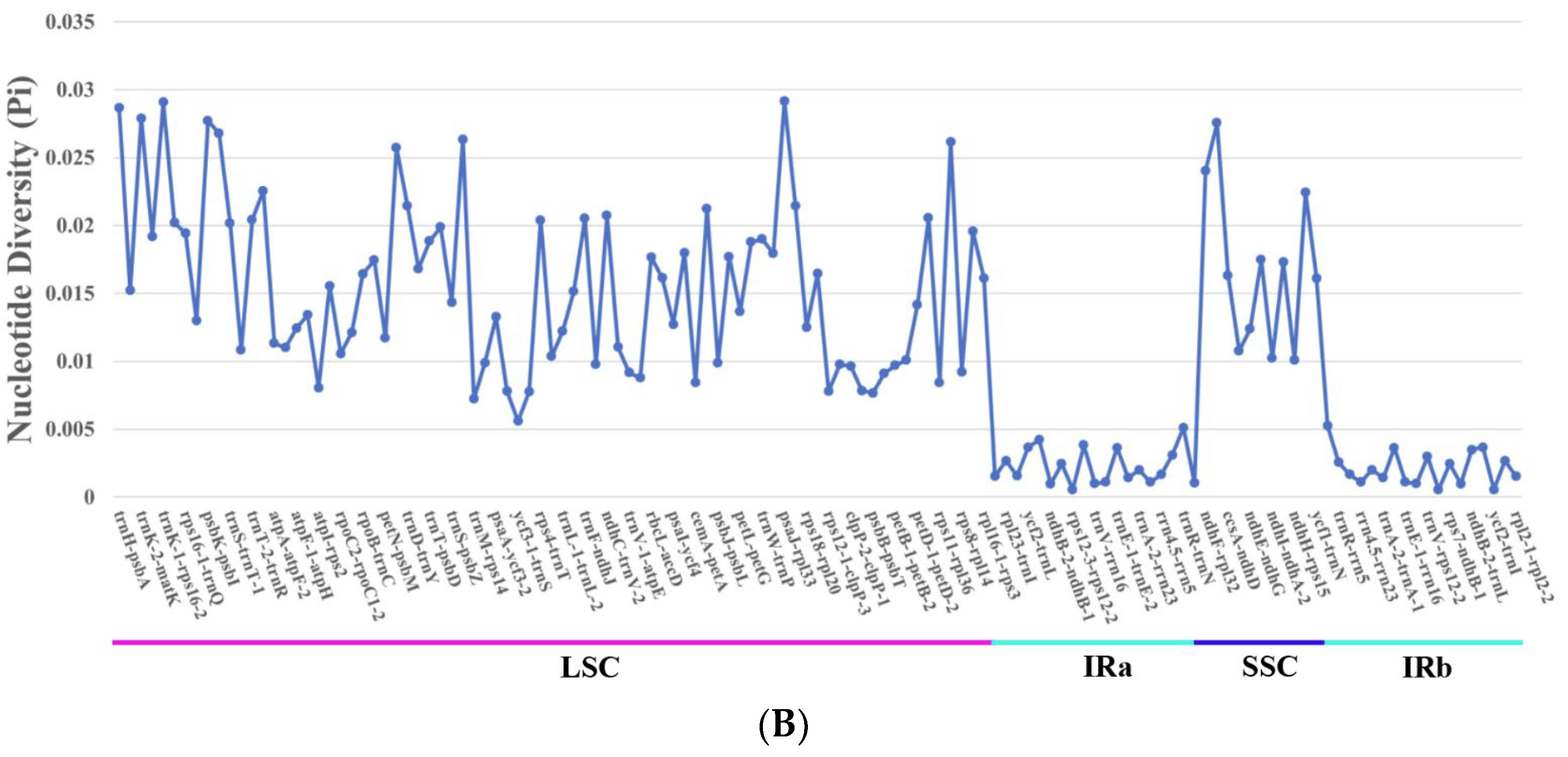
3.7. Nucleotide Diversity
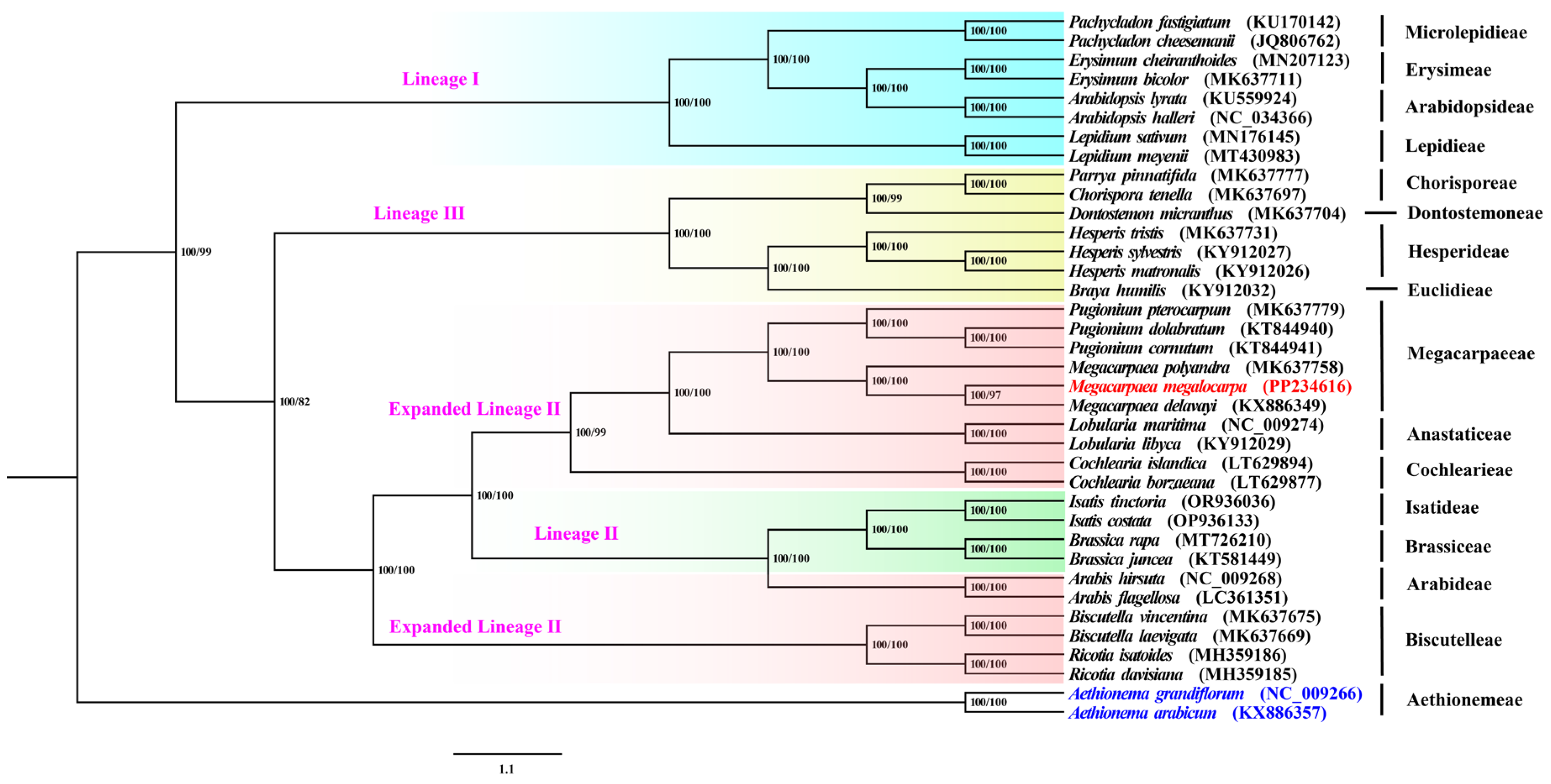
3.8. Phylogenetic Analyses and Estimation of Divergence Times
4. Discussion
4.1. Architecture of Chloroplast Genomes in Megacarpaeeae
4.2. Phylogeny of Chloroplast Genome of Megacarpaeeae
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Al-Shehbaz, I.A. A Generic and Tribal Synopsis of the Brassicaceae (Cruciferae). Taxon 2012, 61, 931–954. [Google Scholar] [CrossRef]
- Nikolov, L.A.; Shushkov, P.; Nevado, B.; Gan, X.; Al-Shehbaz, I.A.; Filatov, D.; Bailey, C.D.; Tsiantis, M. Resolving the Backbone of the Brassicaceae Phylogeny for Investigating Trait Diversity. New Phytol. 2019, 222, 1638–1651. [Google Scholar] [CrossRef]
- Shankar, S.; Segaran, G.; Sundar, R.D.V.; Settu, S.; Sathiavelu, M. Brassicaceae—A Classical Review on Its Pharmacological Activities. Int. J. Pharm. Sci. Rev. Res. 2019, 55, 107–113. [Google Scholar]
- Higdon, J.; Delage, B.; Williams, D.; Dashwood, R. Cruciferous Vegetables and Human Cancer Risk: Epidemiologic Evidence and Mechanistic Basis. Pharmacol. Res. 2007, 55, 224–236. [Google Scholar] [CrossRef]
- Murillo, G.; Mehta, R.G. Cruciferous Vegetables and Cancer Prevention. Nutr. Cancer 2001, 41, 17–28. [Google Scholar] [CrossRef]
- Ilahy, R.; Tlili, I.; Pék, Z.; Montefusco, A.; Siddiqui, M.W.; Homa, F.; Hdider, C.; R’Him, T.; Lajos, H.; Lenucci, M.S. Pre-and Post-Harvest Factors Affecting Glucosinolate Content in Broccoli. Front. Nutr. 2020, 7, 147. [Google Scholar] [CrossRef]
- Guo, X.; Liu, J.; Hao, G.; Zhang, L.; Mao, K.; Wang, X.; Zhang, D.; Ma, T.; Hu, Q.; Al-Shehbaz, I.A.; et al. Plastome Phylogeny and Early Diversification of Brassicaceae. BMC Genom. 2017, 18, 176. [Google Scholar] [CrossRef]
- Huang, C.-H.; Sun, R.; Hu, Y.; Zeng, L.; Zhang, N.; Cai, L.; Zhang, Q.; Koch, M.A.; Al-Shehbaz, I.; Edger, P.P.; et al. Resolution of Brassicaceae Phylogeny Using Nuclear Genes Uncovers Nested Radiations and Supports Convergent Morphological Evolution. Mol. Biol. Evol. 2016, 33, 394–412. [Google Scholar] [CrossRef]
- Hendriks, K.P.; Kiefer, C.; Al-Shehbaz, I.A.; Bailey, C.D.; Van Huysduynen, A.H.; Nikolov, L.A.; Nauheimer, L.; Zuntini, A.R.; German, D.A.; Franzke, A. Global Brassicaceae Phylogeny Based on Filtering of 1,000-Gene Dataset. Curr. Biol. 2023, 33, 4052–4068. [Google Scholar] [CrossRef]
- Cheo, T.; Lu, L.; Yang, G.; Al-Shehbaz, I.; Dorofeev, V. Flora of China; Science Press: Beijing, China, 2001; Volume 8, pp. 39–40. [Google Scholar]
- Shen, L.; Liu, X.; Shi, G.; Yang, Y.; Li, B. Effect of Megacarpaea delavayi Franch on Digestive Juice in Rat with Heat Due to Food Stagnation. Chin. J. Ethnomed. Ethnopharm. 2009, 18, 1–3. [Google Scholar]
- Singh, A.; Nautiyal, M.C.; Curti, R.N.; Fenu, G. The Phenological Growth Stages of Megacarpaea polyandra Benth. Ex Madden: A High Valued Traditional Medicinal Plant of the Himalaya. Genet. Resour. Crop Evol. 2022, 69, 431–441. [Google Scholar] [CrossRef]
- Kiefer, M.; Schmickl, R.; German, D.A.; Mandáková, T.; Lysak, M.A.; Al-Shehbaz, I.A.; Franzke, A.; Mummenhoff, K.; Stamatakis, A.; Koch, M.A. BrassiBase: Introduction to a Novel Knowledge Database on Brassicaceae Evolution. Plant Cell Physiol. 2014, 55, e3. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I. Chloroplast Genome Sequencing: Some Reflections. Next Gen. Seq. Appl. 2015, 2, 1000119. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, F.; Yang, D.-G.; Li, W.; Zhou, X.-J.; Pei, X.-Y.; Liu, Y.-G.; He, K.-L.; Zhang, W.-S.; Ren, Z.-Y. Comparative Chloroplast Genomics of Gossypium Species: Insights into Repeat Sequence Variations and Phylogeny. Front. Plant Sci. 2018, 9, 376. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, W.; Xiang, K.; Wu, Z.; Gu, C. Advancements in Plant Phylogenomics in the Genomic Era. J. Zhejiang AF Univ. 2023, 40, 227–236. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; de Pamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A Fast and Versatile Toolkit for Accurate de Novo Assembly of Organelle Genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an Integrated Plastome Sequence Annotator and Analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A Suite of Tools for Generating Physical Maps of Plastid and Mitochondrial Genomes and Visualizing Expression Data Sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST Databases for the Development and Characterization of Gene-Derived SSR-Markers in Barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S. REPuter: The Manifold Applications of Repeat Analysis on a Genomic Scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An Online Program to Visualize the Junction Sites of Chloroplast Genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Peden, J. Analysis of Codon Usage. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 2000. [Google Scholar]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational Tools for Comparative Genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinf. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A Toolkit Incorporating γ-Series Methods and Sliding Window Strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A Software for Comprehensive Analysis of DNA Polymorphism Data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An Integrated and Scalable Desktop Platform for Streamlined Molecular Sequence Data Management and Evolutionary Phylogenetics Studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef]
- Hohmann, N.; Wolf, E.M.; Lysak, M.A.; Koch, M.A. A Time-Calibrated Road Map of Brassicaceae Species Radiation and Evolutionary History. Plant Cell 2015, 27, 2770–2784. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Hu, H.; Guo, X.; Ma, Y.; Liu, J.; Ma, T. Characterization of the Complete Chloroplast Genome of Two Sister Species of Pugionium (Brassicaceae). Conserv. Genet. Resour. 2016, 8, 243–245. [Google Scholar] [CrossRef]
- Javaid, N.; Ramzan, M.; Khan, I.A.; Alahmadi, T.A.; Datta, R.; Fahad, S.; Danish, S. The Chloroplast Genome of Farsetia hamiltonii Royle, Phylogenetic Analysis, and Comparative Study with Other Members of Clade C of Brassicaceae. BMC Plant Biol. 2022, 22, 384. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Zhao, L.; Xu, T.; Li, C.; Shen, R. The Complete Chloroplast Genome of Lepidium Latifolium linnaeus and Phylogenetic Analysis of Brassicaceae. Mitochondrial DNA B 2021, 6, 178–179. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Zeng, T.; Feng, Q.; Hu, L.; Luo, X.; Weng, Q.; He, J.; Zhu, B. The Complete Chloroplast Genome Sequence of Yellow Mustard (Sinapis alba L.) and Its Phylogenetic Relationship to Other Brassicaceae Species. Gene 2020, 731, 144340. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, R.; Li, J.; Zheng, K.; Xiao, J.; Zheng, Y. Analyses of Chloroplast Genome of Eutrema japonicum Provide New Insights into the Evolution of Eutrema Species. Agronomy 2021, 11, 2546. [Google Scholar] [CrossRef]
- Mann, S.; Chen, Y.-P.P. Bacterial Genomic G+C Composition-Eliciting Environmental Adaptation. Genomics 2010, 95, 7–15. [Google Scholar] [CrossRef]
- Ebert, D.; Peakall, R. Chloroplast Simple Sequence Repeats (cpSSRs): Technical Resources and Recommendations for Expanding cpSSR Discovery and Applications to a Wide Array of Plant Species. Mol. Ecol. Resour. 2009, 9, 673–690. [Google Scholar] [CrossRef] [PubMed]
- George, B.; Bhatt, B.S.; Awasthi, M.; George, B.; Singh, A.K. Comparative Analysis of Microsatellites in Chloroplast Genomes of Lower and Higher Plants. Curr. Genet. 2015, 61, 665–677. [Google Scholar] [CrossRef]
- Kuang, D.-Y.; Wu, H.; Wang, Y.-L.; Gao, L.-M.; Zhang, S.-Z.; Lu, L. Complete Chloroplast Genome Sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA Barcoding and Population Genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef]
- Powell, W.; Morgante, M.; McDevitt, R.; Vendramin, G.G.; Rafalski, J.A. Polymorphic Simple Sequence Repeat Regions in Chloroplast Genomes: Applications to the Population Genetics of Pines. Proc. Natl. Acad. Sci. USA 1995, 92, 7759–7763. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, Y.; Liu, Y.; Xu, C.; Yuan, Q.; Guo, L.; Huang, L. Evolutionary and Phylogenetic Aspects of the Chloroplast Genome of Chaenomeles Species. Sci. Rep. 2020, 10, 11466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, L.; Liu, A.; Chen, J.; Wu, L.; Hu, W.; Zhang, W.; Kim, K.; Lee, S.-C.; Yang, T.-J. The Complete Chloroplast Genome Sequences of Five Epimedium Species: Lights into Phylogenetic and Taxonomic Analyses. Front. Plant Sci. 2016, 7, 306. [Google Scholar] [CrossRef]
- Wei, S.; Liufu, Y.; Zheng, H.; Chen, H.; Lai, Y.; Liu, Y.; Ye, Q.; Tang, S. Using Phylogenomics to Untangle the Taxonomic Incongruence of Yellow-flowered Camellia Species (Theaceae) in China. J. Syst. Evol. 2023, 61, 748–763. [Google Scholar] [CrossRef]
- Wei, R.; Li, Q. The Complete Chloroplast Genome of Endangered Species Stemona parviflora: Insight into the Phylogenetic Relationship and Conservation Implications. Genes 2022, 13, 1361. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Messing, J. High-Throughput Sequencing of Three Lemnoideae (Duckweeds) Chloroplast Genomes from Total DNA. PLoS ONE 2011, 6, e24670. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-B.; Yang, S.-X.; Li, H.-T.; Yang, J.; Li, D.-Z. Comparative Chloroplast Genomes of Camellia Species. PLoS ONE 2013, 8, e73053. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative Analysis of the Complete Chloroplast Genomes of Five Quercus Species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef]
- Zhou, T.; Zhu, H.; Wang, J.; Xu, Y.; Xu, F.; Wang, X. Complete Chloroplast Genome Sequence Determination of Rheum Species and Comparative Chloroplast Genomics for the Members of Rumiceae. Plant Cell Rep. 2020, 39, 811–824. [Google Scholar] [CrossRef]
- Kikuchi, S.; Bédard, J.; Hirano, M.; Hirabayashi, Y.; Oishi, M.; Imai, M.; Takase, M.; Ide, T.; Nakai, M. Uncovering the Protein Translocon at the Chloroplast Inner Envelope Membrane. Science 2013, 339, 571–574. [Google Scholar] [CrossRef]
- Li, L.; Hu, Y.; He, M.; Zhang, B.; Wu, W.; Cai, P.; Huo, D.; Hong, Y. Comparative Chloroplast Genomes: Insights into the Evolution of the Chloroplast Genome of Camellia sinensis and the Phylogeny of Camellia. BMC Genom. 2021, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Gautier, C. Codon Usage in Bacteria: Correlation with Gene Expressivity. Nucleic Acids Res. 1982, 10, 7055–7074. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, L.; Xu, W.; Huo, Z.; Yang, P.; Zhang, Q.; Wang, H.; Li, P.; Lu, X. The Complete Chloroplast Genome Sequence of Cyathula officinalis and Comparative Analysis with Four Related Species. Gene 2022, 839, 146728. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, Z.; Li, X.; Malik, K.; Li, C. Comparative Analyses of Complete Chloroplast Genomes of Microula sikkimensis and Related Species of Boraginaceae. Genes 2024, 15, 226. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Gao, Z.; Hu, L.; Wang, H. Characteristics of the Chloroplast Genome and Phylogenetic Studies of Crambe abyssinica. Biotechnol. Bull. 2022, 38, 174. [Google Scholar] [CrossRef]
- Dong, W.-L.; Wang, R.-N.; Zhang, N.-Y.; Fan, W.-B.; Fang, M.-F.; Li, Z.-H. Molecular Evolution of Chloroplast Genomes of Orchid Species: Insights into Phylogenetic Relationship and Adaptive Evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Cheng, L.; Huang, W.; Cao, Q.; Zhou, L.; Jia, W.; Lan, Y. Chloroplast Genomes of Seven Species of Coryloideae (Betulaceae): Structures and Comparative Analysis. Genome 2020, 63, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Dos Reis, M. How to Calculate the Non-Synonymous to Synonymous Rate Ratio of Protein-Coding Genes under the Fisher-Wright Mutation-Selection Framework. Biol. Lett. 2015, 11, 20141031. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Estimating Synonymous and Nonsynonymous Substitution Rates under Realistic Evolutionary Models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef]
- Yu, T.; Gao, J.; Liao, P.-C.; Li, J.-Q.; Ma, W.-B. Insights into Comparative Analyses and Phylogenomic Implications of Acer (Sapindaceae) Inferred from Complete Chloroplast Genomes. Front. Genet. 2022, 12, 791628. [Google Scholar] [CrossRef]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; de Pamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J.; et al. Methods for Obtaining and Analyzing Whole Chloroplast Genome Sequences. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2005; Volume 395, pp. 348–384. ISBN 978-0-12-182800-4. [Google Scholar] [CrossRef]
- Liu, L.; Du, X.; Guo, C.; Li, D. Resolving Robust Phylogenetic Relationships of Core Brassicaceae Using Genome Skimming Data. J. Syst. Evol. 2021, 59, 442–453. [Google Scholar] [CrossRef]
- Couvreur, T.L.P.; Franzke, A.; Al-Shehbaz, I.A.; Bakker, F.T.; Koch, M.A.; Mummenhoff, K. Molecular Phylogenetics, Temporal Diversification, and Principles of Evolution in the Mustard Family (Brassicaceae). Mol. Biol. Evol. 2010, 27, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, T. The Age of the Taklimakan Desert. Science 2006, 312, 1621. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Zhang, M.; Peng, D.; Wang, H.; Wu, M.; Chen, R. Tectonic and Climatic Pattern Evolution of the Middle-Cenozoic in Northwest China. Mar. Geol. Quat. Geol. 2013, 33, 153–168. [Google Scholar] [CrossRef]
- Li, X.; Dong, G. Discussion on the Formation Era and Causes of Arid Environment in Northwest China. Quat. Sci. 2006, 26, 895–904. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length (bp) | GC Content (%) | Number of Gene | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genome | LSC | IR | SSC | Total | LSC | IR | SSC | Total | Protein Coding | tRNA | rRNA | |
| M. megalocarpa | 154,877 | 84,008 | 26,446 | 17,977 | 36.20 | 33.96 | 42.31 | 29.08 | 129 | 85 | 36 | 8 |
| M. delavayi | 154,401 | 83,704 | 26,336 | 18,025 | 36.20 | 33.89 | 42.34 | 28.98 | 130 | 85 | 37 | 8 |
| M. polyandra | 154,949 | 83,991 | 26,433 | 18,092 | 36.10 | 33.85 | 42.28 | 28.91 | 132 | 87 | 37 | 8 |
| P. cornutum | 154,992 | 84,104 | 26,437 | 18,014 | 36.20 | 33.92 | 42.36 | 29.05 | 130 | 85 | 37 | 8 |
| P. dolabratum | 155,022 | 84,097 | 26,437 | 18,031 | 36.20 | 33.93 | 42.36 | 29.03 | 130 | 85 | 37 | 8 |
| P. pterocarpum | 154,534 | 83,762 | 26,442 | 17,888 | 36.30 | 33.92 | 42.36 | 29.16 | 132 | 87 | 37 | 8 |
| Category | Gene Group | Gene Name |
|---|---|---|
| Photosynthesis | Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbG, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunits of NADH dehydrogenase | ndhA*, ndhB*(2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ | |
| Subunits of cytochrome b/f complex | petA, petB*, petD*, petG, petL, petN | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF*, atpH, atpI | |
| Large subunit of rubisco | rbcL | |
| Self-replication | Proteins of large ribosomal subunit | rbcL |
| Proteins of small ribosomal subunit | rps11, rps12**(2), rps14, rps15, rps16*, rps18, rps19, rps2, rps3, rps4, rps7(2), rps8 | |
| Subunits of RNA polymerase | rpoA, rpoB, rpoC1*, rpoC2 | |
| Ribosomal RNAs | rrn16(2), rrn23(2), rrn4.5(2), rrn5(2) | |
| Transfer RNAs | trnA-UGC*(2), trnC-GCA, trnD-GUC, trnE-UUC, trnE-UUC*(2), trnF-GAA, trnG-GCC, trnH-GUG, trnI-CAU(2), trnK-UUU*, trnL-CAA(2), trnL-UAA*, trnL-UAG, trnM-CAU, trnN-GUU(2), trnP-UGG, trnQ-UUG, trnR-ACG(2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-CGU*, trnT-GGU, trnT-UGU, trnV-GAC(2), trnV-UAC*, trnW-CCA, trnY-GUA | |
| Other genes | Maturase | matK |
| Protease | clpP** | |
| Envelope membrane protein | cemA | |
| Acetyl-CoA carboxylase | accD | |
| c-type cytochrome synthesis gene | ccsA | |
| Genes of unknown function | Conserved hypothetical chloroplast ORF | ycf1(2), ycf2(2), ycf3*, ycf4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Shi, X.; Tian, H.; Qiu, J.; Ma, H.; Tan, D. Complete Chloroplast Genome of Megacarpaea megalocarpa and Comparative Analysis with Related Species from Brassicaceae. Genes 2024, 15, 886. https://doi.org/10.3390/genes15070886
Zhang Z, Shi X, Tian H, Qiu J, Ma H, Tan D. Complete Chloroplast Genome of Megacarpaea megalocarpa and Comparative Analysis with Related Species from Brassicaceae. Genes. 2024; 15(7):886. https://doi.org/10.3390/genes15070886
Chicago/Turabian StyleZhang, Zhuo, Xiaojun Shi, Haowen Tian, Juan Qiu, Hanze Ma, and Dunyan Tan. 2024. "Complete Chloroplast Genome of Megacarpaea megalocarpa and Comparative Analysis with Related Species from Brassicaceae" Genes 15, no. 7: 886. https://doi.org/10.3390/genes15070886