

Revealing Differential RNA Editing Specificity of Human ADAR1 and ADAR2 in Schizosaccharomyces pombe
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
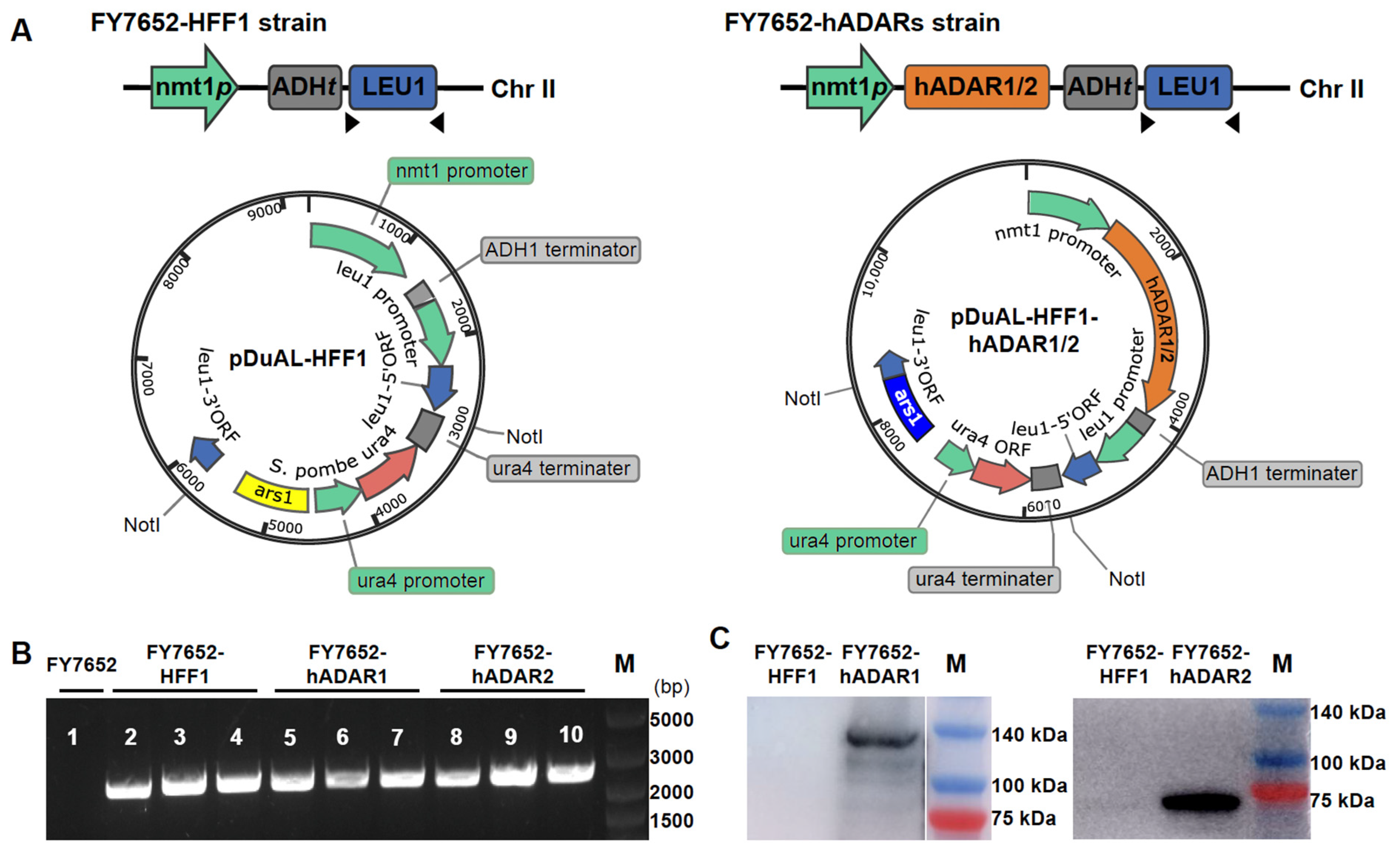
2.1. Construction of hADAR1 and hADAR2 Expression Vectors
2.2. S. pombe Strains and Transformation
2.3. Western Blotting
2.4. S. pombe Strains Cultivation
2.5. Transcriptome Sequencing (mRNA-Seq)
2.6. Pipeline for Identification of A-to-I RNA Editing Events
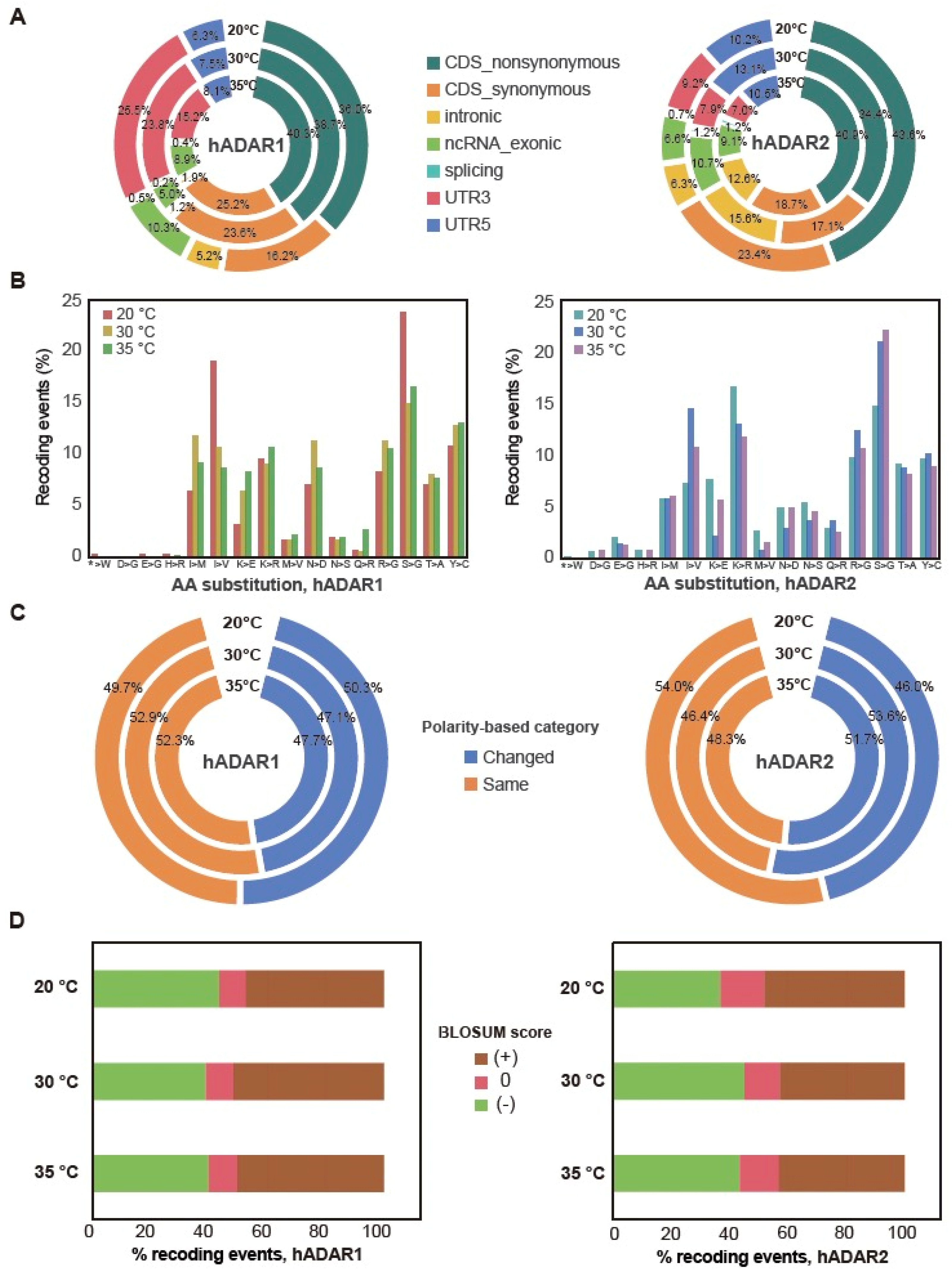
2.7. Gene-Based Annotation of A-to-I RNA Editing Sites
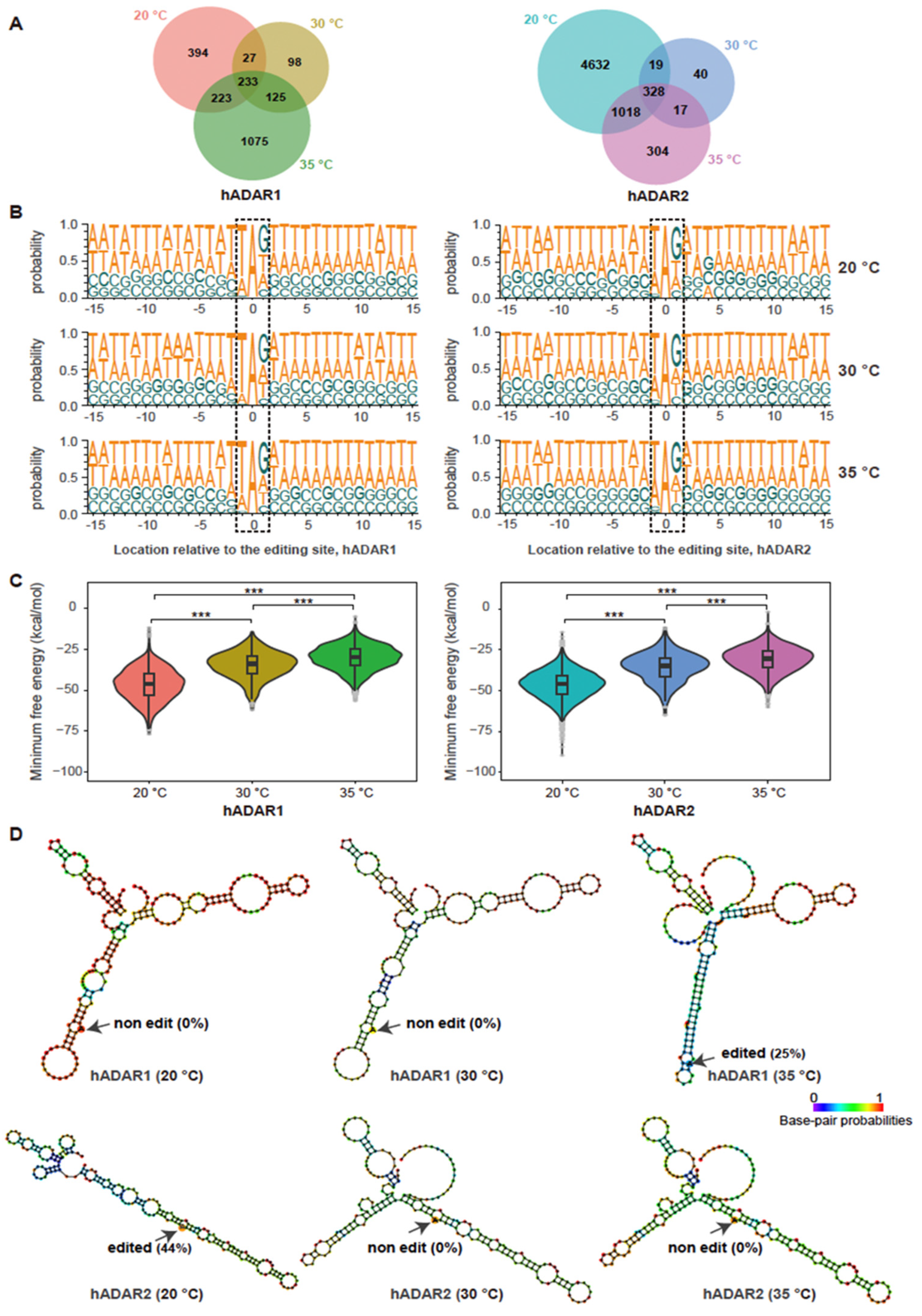
2.8. Sequence Logo Analysis of Nucleotides Neighboring the Detected Editing Sites
2.9. Analysis of Minimum Free Energy for Secondary RNA Structure at Editing Sites
3. Results
3.1. Construction of RNA Editing Machinery in S. pombe
3.2. hADAR1 and hADAR2 Displaying Distinct RNA Editing Specificity in S. pombe
3.3. Varying Temperature Affecting RNA Editing by hADAR1 and hADAR2 in S. pombe
3.4. Genes’ Functions Possibly Affected by the Editing of Their Transcript
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bass, B.L.; Weintraub, H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 1988, 55, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, G.; Lin, W.; Piskol, R.; Tan, M.H.; Davis, C.; Li, J.B. Accurate identification of human Alu and non-Alu RNA editing sites. Nat. Methods 2012, 9, 579–581. [Google Scholar] [CrossRef]
- Huang, H.; Tan, B.Z.; Shen, Y.; Tao, J.; Jiang, F.; Sung, Y.Y.; Ng, C.K.; Raida, M.; Köhr, G.; Higuchi, M.; et al. RNA editing of the IQ domain in Ca(v)1.3 channels modulates their Ca2+-dependent inactivation. Neuron 2012, 73, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Rueter, S.M.; Dawson, T.R.; Emeson, R.B. Regulation of alternative splicing by RNA editing. Nature 1999, 399, 75–80. [Google Scholar] [CrossRef]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef]
- Liddicoat, B.J.; Piskol, R.; Chalk, A.M.; Ramaswami, G.; Higuchi, M.; Hartner, J.C.; Li, J.B.; Seeburg, P.H.; Walkley, C.R. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015, 349, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Forni, D.; Mozzi, A.; Pontremoli, C.; Vertemara, J.; Pozzoli, U.; Biasin, M.; Bresolin, N.; Clerici, M.; Cagliani, R.; Sironi, M. Diverse selective regimes shape genetic diversity at ADAR genes and at their coding targets. RNA Biol. 2015, 12, 149–161. [Google Scholar] [CrossRef]
- Palladino, M.J.; Keegan, L.P.; O’Connell, M.A.; Reenan, R.A. dADAR, a Drosophila double-stranded RNA-specific adenosine deaminase is highly developmentally regulated and is itself a target for RNA editing. RNA 2000, 6, 1004–1018. [Google Scholar] [CrossRef]
- Albertin, C.B.; Simakov, O.; Mitros, T.; Wang, Z.Y.; Pungor, J.R.; Edsinger-Gonzales, E.; Brenner, S.; Ragsdale, C.W.; Rokhsar, D.S. The octopus genome and the evolution of cephalopod neural and morphological novelties. Nature 2015, 524, 220–224. [Google Scholar] [CrossRef]
- Huntley, M.A.; Lou, M.; Goldstein, L.D.; Lawrence, M.; Dijkgraaf, G.J.; Kaminker, J.S.; Gentleman, R. Complex regulation of ADAR-mediated RNA-editing across tissues. BMC Genom. 2016, 17, 61. [Google Scholar] [CrossRef]
- Patterson, J.B.; Samuel, C.E. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: Evidence for two forms of the deaminase. Mol. Cell. Biol. 1995, 15, 5376–5388. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.X.; Cho, D.S.; Wang, Q.; Lai, F.; Carter, K.C.; Nishikura, K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 2000, 6, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Costa Cruz, P.H.; Kato, Y.; Nakahama, T.; Shibuya, T.; Kawahara, Y. A comparative analysis of ADAR mutant mice reveals site-specific regulation of RNA editing. RNA 2020, 26, 454–469. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.H.; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; Ariyoshi, K.; et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 2017, 550, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Chalk, A.M.; Taylor, S.; Heraud-Farlow, J.E.; Walkley, C.R. The majority of A-to-I RNA editing is not required for mammalian homeostasis. Genome Biol. 2019, 20, 268. [Google Scholar] [CrossRef] [PubMed]
- Eifler, T.; Pokharel, S.; Beal, P.A. RNA-Seq analysis identifies a novel set of editing substrates for human ADAR2 present in Saccharomyces cerevisiae. Biochemistry 2013, 52, 7857–7869. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Park, S.; Beal, P.A. Selective Recognition of RNA Substrates by ADAR Deaminase Domains. Biochemistry 2018, 57, 1640–1651. [Google Scholar] [CrossRef]
- Dawson, T.R.; Sansam, C.L.; Emeson, R.B. Structure and sequence determinants required for the RNA editing of ADAR2 substrates. J. Biol. Chem. 2004, 279, 4941–4951. [Google Scholar] [CrossRef]
- Duan, Y.; Dou, S.; Luo, S.; Zhang, H.; Lu, J. Adaptation of A-to-I RNA editing in Drosophila. PLoS Genet. 2017, 13, e1006648. [Google Scholar] [CrossRef]
- Lehmann, K.A.; Bass, B.L. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry 2000, 39, 12875–12884. [Google Scholar] [CrossRef]
- Eggington, J.M.; Greene, T.; Bass, B.L. Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2011, 2, 319. [Google Scholar] [CrossRef] [PubMed]
- Liscovitch-Brauer, N.; Alon, S.; Porath, H.T.; Elstein, B.; Unger, R.; Ziv, T.; Admon, A.; Levanon, E.Y.; Rosenthal, J.J.C.; Eisenberg, E. Trade-off between Transcriptome Plasticity and Genome Evolution in Cephalopods. Cell 2017, 169, 191–202.e11. [Google Scholar] [CrossRef] [PubMed]
- Zambrano-Mila, M.S.; Witzenberger, M.; Rosenwasser, Z.; Uzonyi, A.; Nir, R.; Ben-Aroya, S.; Levanon, E.Y.; Schwartz, S. Dissecting the basis for differential substrate specificity of ADAR1 and ADAR2. Nat. Commun. 2023, 14, 8212. [Google Scholar] [CrossRef]
- Bass, B.L. RNA editing and hypermutation by adenosine deamination. Trends Biochem. Sci. 1997, 22, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Källman, A.M.; Sahlin, M.; Ohman, M. ADAR2 A-->I editing: Site selectivity and editing efficiency are separate events. Nucleic Acids Res. 2003, 31, 4874–4881. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, Y.; Yamashita, T.; Hideyama, T.; Tsuji, S.; Suzuki, N.; Kwak, S. Determination of editors at the novel A-to-I editing positions. Neurosci. Res. 2008, 61, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, A.; Shirai, A.; Yashiroda, Y.; Kamata, A.; Horinouchi, S.; Yoshida, M. pDUAL, a multipurpose, multicopy vector capable of chromosomal integration in fission yeast. Yeast 2004, 21, 1289–1305. [Google Scholar] [CrossRef] [PubMed]
- Sabatinos, S.A.; Forsburg, S.L. Molecular genetics of Schizosaccharomyces pombe. Methods Enzymol. 2010, 470, 759–795. [Google Scholar]
- Zhang, N.; Jing, X.; Liu, Y.; Chen, M.; Zhu, X.; Jiang, J.; Wang, H.; Li, X.; Hao, P. Interfering with retrotransposition by two types of CRISPR effectors: Cas12a and Cas13a. Cell Discov. 2020, 6, 30. [Google Scholar] [CrossRef]
- Jing, X.; Xie, B.; Chen, L.; Zhang, N.; Jiang, Y.; Qin, H.; Wang, H.; Hao, P.; Yang, S.; Li, X. Implementation of the CRISPR-Cas13a system in fission yeast and its repurposing for precise RNA editing. Nucleic Acids Res. 2018, 46, e90. [Google Scholar] [CrossRef]
- Chen, L.; Ou, L.; Jing, X.; Kong, Y.; Xie, B.; Zhang, N.; Shi, H.; Qin, H.; Li, X.; Hao, P. DeepEdit: Single-molecule detection and phasing of A-to-I RNA editing events using nanopore direct RNA sequencing. Genome Biol. 2023, 24, 75. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhou, H.; Kong, Y.; Pan, B.; Chen, L.; Wang, H.; Hao, P.; Li, X. The Landscape of A-to-I RNA Editome Is Shaped by Both Positive and Purifying Selection. PLoS Genet. 2016, 12, e1006191. [Google Scholar] [CrossRef]
- Xu, H.; Luo, X.; Qian, J.; Pang, X.; Song, J.; Qian, G.; Chen, J.; Chen, S. FastUniq: A fast de novo duplicates removal tool for paired short reads. PLoS ONE 2012, 7, e52249. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Buchumenski, I.; Bartok, O.; Ashwal-Fluss, R.; Pandey, V.; Porath, H.T.; Levanon, E.Y.; Kadener, S. Dynamic hyper-editing underlies temperature adaptation in Drosophila. PLoS Genet. 2017, 13, e1006931. [Google Scholar] [CrossRef]
- Birk, M.A.; Liscovitch-Brauer, N.; Dominguez, M.J.; McNeme, S.; Yue, Y.; Hoff, J.D.; Twersky, I.; Verhey, K.J.; Sutton, R.B.; Eisenberg, E.; et al. Temperature-dependent RNA editing in octopus extensively recodes the neural proteome. Cell 2023, 186, 2544–2555.e2513. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Qu, K.; Ouyang, Z.; Kertesz, M.; Li, J.; Tibshirani, R.; Makino, D.L.; Nutter, R.C.; Segal, E.; Chang, H.Y. Genome-wide measurement of RNA folding energies. Mol. Cell 2012, 48, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Gerber, A.; Grosjean, H.; Melcher, T.; Keller, W. Tad1p, a yeast tRNA-specific adenosine deaminase, is related to the mammalian pre-mRNA editing enzymes ADAR1 and ADAR2. EMBO J. 1998, 17, 4780–4789. [Google Scholar] [CrossRef] [PubMed]
- Kuttan, A.; Bass, B.L. Mechanistic insights into editing-site specificity of ADARs. Proc. Natl. Acad. Sci. USA 2012, 109, E3295–E3304. [Google Scholar] [CrossRef] [PubMed]
- Matthews, M.M.; Thomas, J.M.; Zheng, Y.; Tran, K.; Phelps, K.J.; Scott, A.I.; Havel, J.; Fisher, A.J.; Beal, P.A. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol. 2016, 23, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, H.G.; Beal, P.A. Chemical Modifications in RNA: Elucidating the Chemistry of dsRNA-Specific Adenosine Deaminases (ADARs). Acc. Chem. Res. 2023, 56, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.M.; Beal, P.A. How do ADARs bind RNA? New protein-RNA structures illuminate substrate recognition by the RNA editing ADARs. BioEssays 2017, 39, 1600187. [Google Scholar] [CrossRef] [PubMed]
- Stefl, R.; Oberstrass, F.C.; Hood, J.L.; Jourdan, M.; Zimmermann, M.; Skrisovska, L.; Maris, C.; Peng, L.; Hofr, C.; Emeson, R.B.; et al. The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell 2010, 143, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Riemondy, K.A.; Gillen, A.E.; White, E.A.; Bogren, L.K.; Hesselberth, J.R.; Martin, S.L. Dynamic temperature-sensitive A-to-I RNA editing in the brain of a heterothermic mammal during hibernation. RNA 2018, 24, 1481–1495. [Google Scholar] [CrossRef]
- Duan, Y.; Tang, X.; Lu, J. Evolutionary driving forces of A-to-I editing in metazoans. Wiley Interdiscip. Rev. RNA 2022, 13, e1666. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar]
- Wood, V.; Gwilliam, R.; Rajandream, M.A.; Lyne, M.; Lyne, R.; Stewart, A.; Sgouros, J.; Peat, N.; Hayles, J.; Baker, S.; et al. The genome sequence of Schizosaccharomyces pombe. Nature 2002, 415, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Alon, S.; Garrett, S.C.; Levanon, E.Y.; Olson, S.; Graveley, B.R.; Rosenthal, J.J.; Eisenberg, E. The majority of transcripts in the squid nervous system are extensively recoded by A-to-I RNA editing. eLife 2015, 4, e05198. [Google Scholar] [CrossRef]
- Garrett, S.; Rosenthal, J.J. RNA editing underlies temperature adaptation in K+ channels from polar octopuses. Science 2012, 335, 848–851. [Google Scholar] [CrossRef]
- Rangan, K.J.; Reck-Peterson, S.L. RNA recoding in cephalopods tailors microtubule motor protein function. Cell 2023, 186, 2531–2543.e11. [Google Scholar] [CrossRef] [PubMed]
- Shoshan, Y.; Liscovitch-Brauer, N.; Rosenthal, J.J.C.; Eisenberg, E. Adaptive Proteome Diversification by Nonsynonymous A-to-I RNA Editing in Coleoid Cephalopods. Mol. Biol. Evol. 2021, 38, 3775–3788. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, N.; Chen, P.; Cui, Z.; Zhou, X.; Hao, C.; Xie, B.; Hao, P.; Ye, B.-C.; Li, X.; Jing, X. Revealing Differential RNA Editing Specificity of Human ADAR1 and ADAR2 in Schizosaccharomyces pombe. Genes 2024, 15, 898. https://doi.org/10.3390/genes15070898
Zhang N, Chen P, Cui Z, Zhou X, Hao C, Xie B, Hao P, Ye B-C, Li X, Jing X. Revealing Differential RNA Editing Specificity of Human ADAR1 and ADAR2 in Schizosaccharomyces pombe. Genes. 2024; 15(7):898. https://doi.org/10.3390/genes15070898
Chicago/Turabian StyleZhang, Niubing, Ping Chen, Zilin Cui, Xiaojuan Zhou, Chenhui Hao, Bingran Xie, Pei Hao, Bang-Ce Ye, Xuan Li, and Xinyun Jing. 2024. "Revealing Differential RNA Editing Specificity of Human ADAR1 and ADAR2 in Schizosaccharomyces pombe" Genes 15, no. 7: 898. https://doi.org/10.3390/genes15070898