

invdup(8)(8q24.13q24.3)—A Complex Alteration and Its Clinical Consequences

 , , ,
, , ,  , , and
, , and
Abstract
1. Introduction
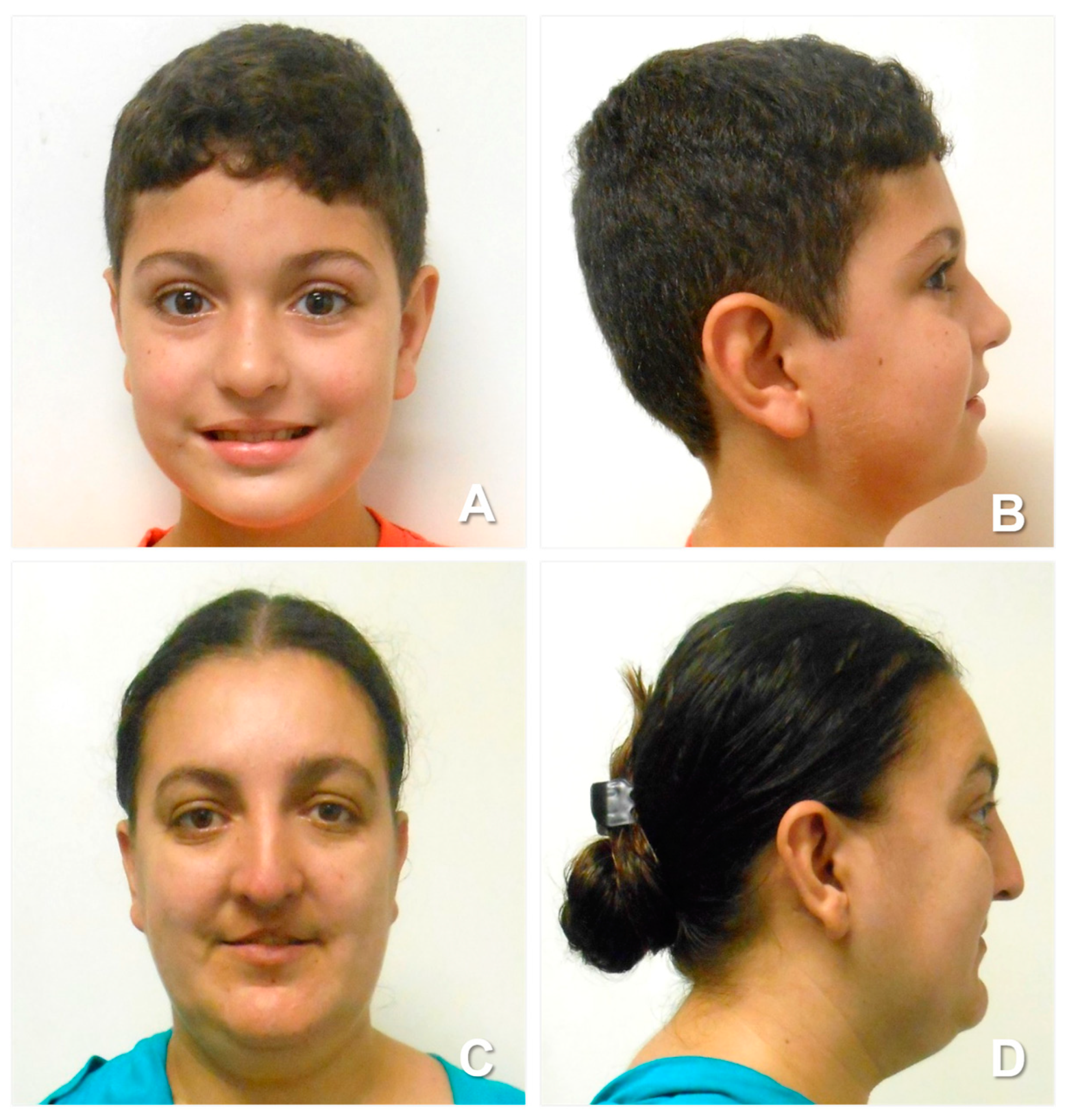
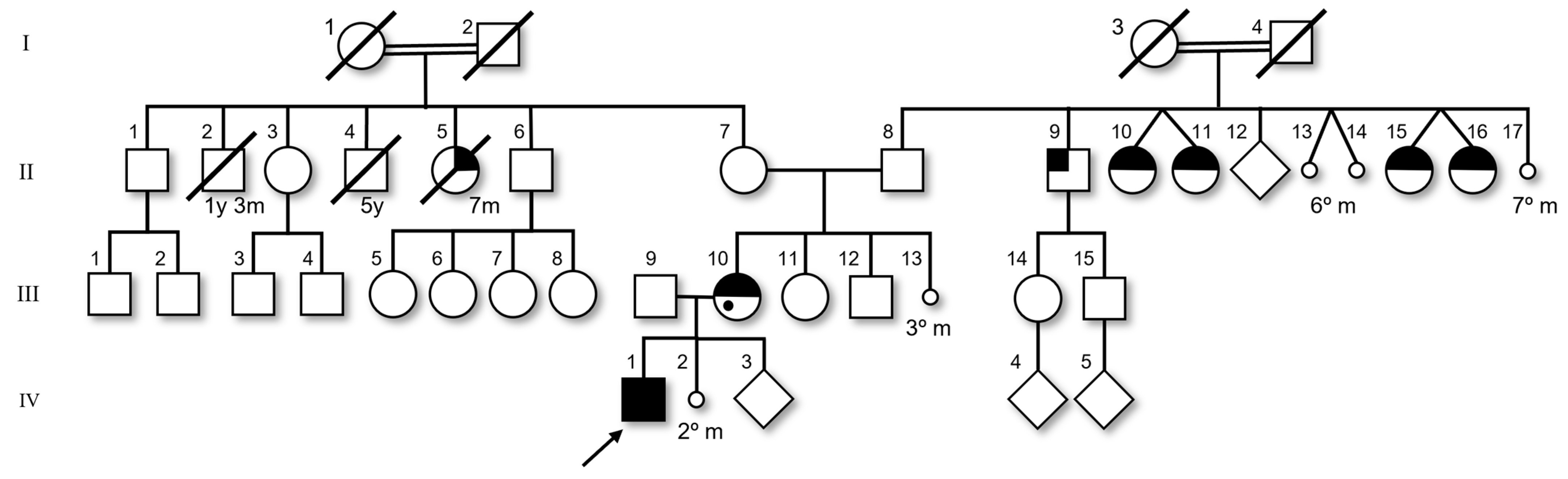
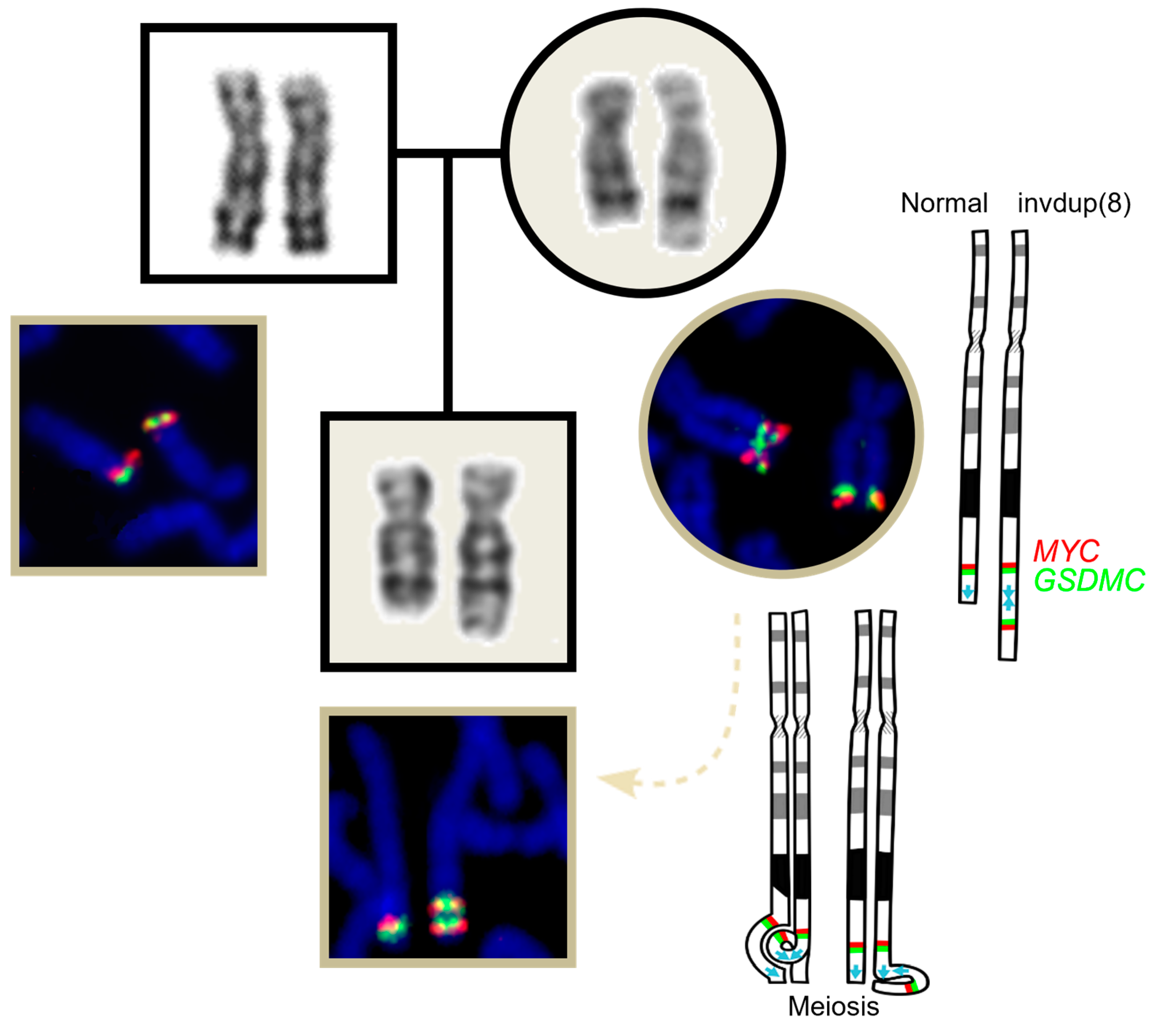
2. Case Report
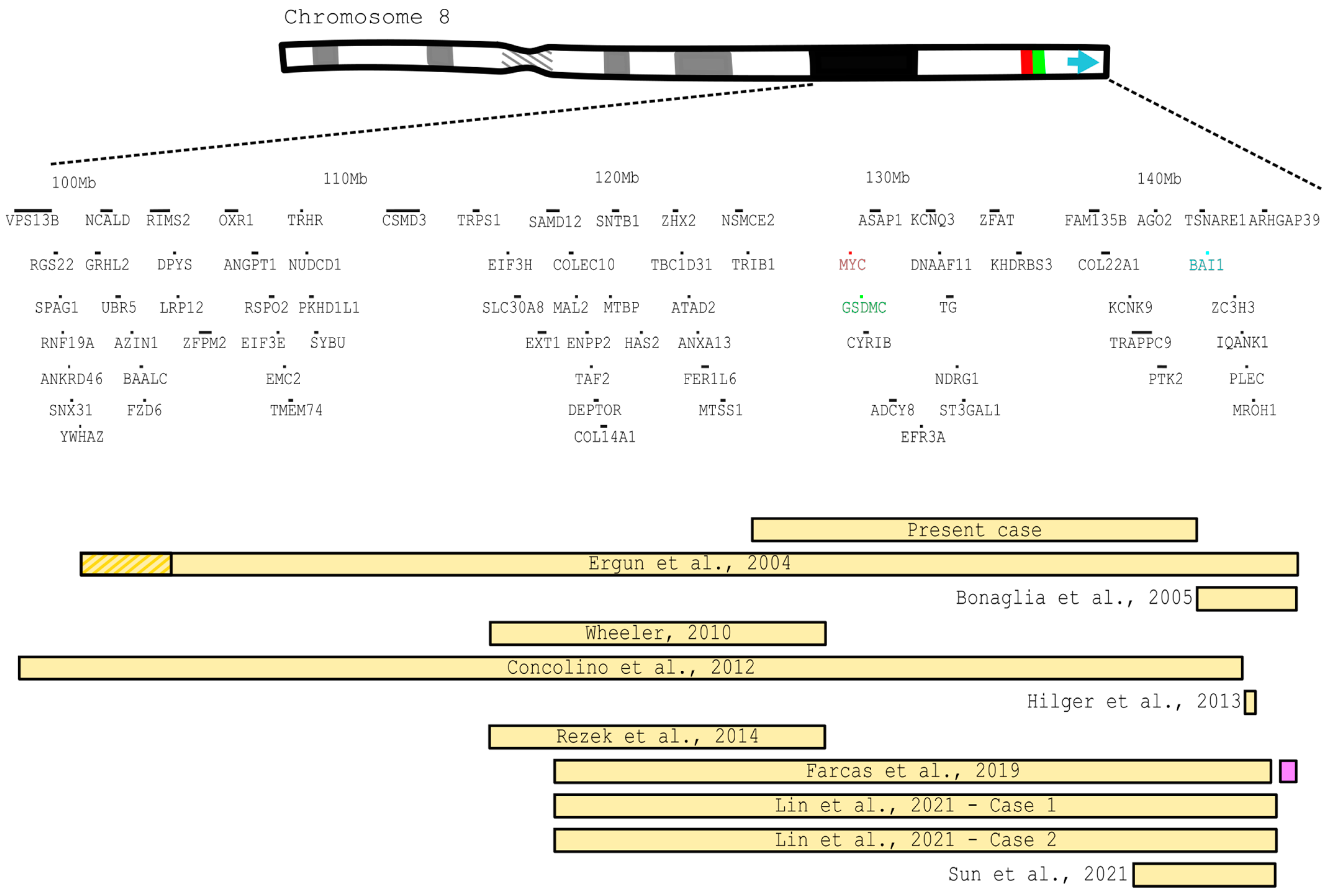
3. Literature Review
4. Discussion
4.1. Facial Features
4.2. Skin Anomalies
4.3. Heart Defects
4.4. Neurologic Aspects
4.5. Miscarriages
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wlasnowolski, M.; Sadowski, M.; Czarnota, T.; Jodkowska, K.; Szalaj, P.; Tang, Z.; Ruan, Y.; Plewczynski, D. 3D-GNOME 2.0: A three-dimensional genome modeling engine for predicting structural variation-driven alterations of chromatin spatial structure in the human genome. Nucleic Acids Res. 2020, 48, W170–W176. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Concolino, D.; Iembo, M.A.; Moricca, M.T.; Rapsomaniki, M.; Marotta, R.; Galesi, O.; Fichera, M.; Romano, C.; Strisciuglio, P. A de novo 8q22.2-24.3 duplication in a patient with mild phenotype. Eur. J. Med. Genet. 2012, 55, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Rezek, R.F.; Rodrigues Abbas, A.A.; Forte Mazzeu, J.; Duarte Miranda, S.M.; Velloso-Rodrigues, C. A rare interstitial duplication of 8q22.1-8q24.3 associated with syndromic bilateral cleft lip/palate. Case Rep. Dent. 2014, 2014, 730375. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pickler, L.; Wilson, R.; Tsai, A.C. Revisiting recombinant 8 syndrome. Am. J. Med. Genet. A. 2011, 155, 1923–1929. [Google Scholar] [CrossRef] [PubMed]
- Holwerda, S.J.; de Laat, W. CTCF: The protein, the binding partners, the binding sites and their chromatin loops. Philos Trans. R Soc. B Biol. Sci. 2013, 368, 20120369. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wechsler, D. Wechsler Intelligence Scale for Children—Third Edition (WISC-III); The Psychological Corporation: San Antonio, TX, USA, 1991. [Google Scholar]
- Riegel, M.; Mergener, R.; Rosa, R.F.; Zen, P. Chromosomal Structural Rearrangements: Characterizing Interstitial Deletions and Duplications in The Clinical Practice. Arch. Pediatr. 2017, JPED-124, 100024. [Google Scholar] [CrossRef]
- Wechsler, D. The Wechsler Intelligence Scale for Children, 4th ed.; Pearson: London, UK, 2003. [Google Scholar]
- Ergun, M.A.; Balci, S.; Konaç, E.; Kan, D.; Menevşe, S.; Bartsch, O. Trisomy of 8q22.3 approximately q23-qter following an unbalanced 1;8 translocation in a boy with multiple anomalies. Turk. J. Pediatr. 2004, 46, 384–387. [Google Scholar] [PubMed]
- Bonaglia, M.C.; Giorda, R.; Tenconi, R.; Pessina, M.; Pramparo, T.; Borgatti, R.; Zuffardi, O. A 2.3 Mb duplication of chromosome 8q24.3 associated with severe mental retardation and epilepsy detected by standard karyotype. Eur. J. Hum. Genet. 2005, 13, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, P.G. 8q23-q24 duplication—Further delineation of a rare chromosomal abnormality. Am. J. Med. Genet. Part A 2010, 152A, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Hilger, A.; Schramm, C.; Pennimpede, T.; Wittler, L.; Dworschak, G.C.; Bartels, E.; Engels, H.; Zink, A.M.; Degenhardt, F.; Müller, A.M.; et al. De novo microduplications at 1q41, 2q37.3, and 8q24.3 in patients with VATER/VACTERL association. Eur. J. Hum. Genet. 2013, 21, 1377–1382. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Farcas, S.; Erdelean, D.; Anne-Elise Szekely, F.; Navolan, D.; Andreescu, N.; Cioca, A. A rare case of partial trisomy 8q24.12-q24.3 and partial monosomy of 8q24.3: Prenatal diagnosis and clinical findings. Taiwan J. Obstet. Gynecol. 2019, 58, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Huang, S.; Ou, X.; Gu, H.; Wang, Y.; Li, P.; Zhou, Y. Mosaic duplication of 8q24.1q24.3 detected by chromosomal microarray but not karyotyping in two unrelated fetuses with cardiac defects. Mol. Cytogenet. 2021, 14, 26. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, H.; Wan, N. Genotype-Phenotype Analysis of 8q24.3 Duplication and 21q22.3 Deletion in a Chinese Patient and Literature Review. Public Health Genom. 2021, 24, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, R.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Iny Stein, T.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Brewer, C.; Holloway, S.; Zawalnyski, P.; Schinzel, A.; FitzPatrick, D. A chromosomal deletion map of human malformations. Am. J. Hum. Genet. 1998, 63, 1153–1159. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Domínguez-Frutos, E.; López-Hernández, I.; Vendrell, V.; Neves, J.; Gallozzi, M.; Gutsche, K.; Quintana, L.; Sharpe, J.; Knoepfler, P.S.; Eisenman, R.N.; et al. N-myc controls proliferation, morphogenesis, and patterning of the inner ear. J. Neurosci. 2011, 31, 7178–7189. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Galupa, R.; Picard, C.; Servant, N.; Nora, E.P.; Zhan, Y.; van Bemmel, J.G.; El Marjou, F.; Johanneau, C.; Borensztein, M.; Ancelin, K.; et al. Inversion of a topological domain leads to restricted changes in its gene expression and affects interdomain communication. Development 2022, 149, dev200568. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Puig, M.; Casillas, S.; Villatoro, S.; Cáceres, M. Human inversions and their functional consequences. Brief. Funct. Genom. 2015, 14, 369–379. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kosuthova, K.; Solc, R. Inversions on human chromosomes. Am. J. Med. Genet. Part A 2023, 191, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, S.; Ludwig, K.U.; Reutter, H.; Herms, S.; Steffens, M.; Rubini, M.; Baluardo, C.; Ferrian, M.; Almeida de Assis, N.; Alblas, M.A.; et al. Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat. Genet. 2009, 41, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, K.U.; Mangold, E.; Herms, S.; Nowak, S.; Reutter, H.; Paul, A.; Becker, J.; Herberz, R.; AlChawa, T.; Nasser, E.; et al. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat. Genet. 2012, 44, 968–971. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pizzo, L.; Jensen, M.; Polyak, A.; Rosenfeld, J.A.; Mannik, K.; Krishnan, A.; McCready, E.; Pichon, O.; Le Caignec, C.; Van Dijck, A.; et al. Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet. Med. 2019, 21, 816–825. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, N.; Yang, T.; Li, J.; Lei, M.; Shi, J.; Qiu, W.; Lian, X. The expression and role of c-Myc in mouse hair follicle morphogenesis and cycling. Acta Histochem. 2012, 114, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Li, C.; Swanson, A.M.; Villalba, R.M.; Guo, J.; Zhang, Z.; Matheny, S.; Murakami, T.; Stephenson, J.R.; Daniel, S.; et al. BAI1 regulates spatial learning and synaptic plasticity in the hippocampus. J. Clin. Investig. 2015, 125, 1497–1508. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shiu, F.H.; Wong, J.C.; Yamamoto, T.; Lala, T.; Purcell, R.H.; Owino, S.; Zhu, D.; Van Meir, E.G.; Hall, R.A.; Escayg, A. Mice lacking full length Adgrb1 (Bai1) exhibit social deficits, increased seizure susceptibility, and altered brain development. Exp. Neurol. 2022, 351, 113994. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Watson, C.T.; Marques-Bonet, T.; Sharp, A.J.; Mefford, H.C. The genetics of microdeletion and microduplication syndromes: An update. Annu. Rev. Genom. Hum. Genet. 2014, 15, 215–244. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, B.G.; Wu, W.J.; Zheng, H.C.; Yang, H.F.; Zuo, Y.X.; Cui, X.P. Long noncoding RNA GAS5 silencing inhibits the expression of KCNQ3 by sponging miR-135a-5p to prevent the progression of epilepsy. Kaohsiung J. Med. Sci. 2019, 35, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Miceli, F.; Soldovieri, M.V.; Weckhuysen, S.; Cooper, E.C.; Taglialatela, M. KCNQ3-Related Disorders. [Updated 28 September 2023]. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 22 May 2014; pp. 1993–2024. [Google Scholar]
- Miceli, F.; Striano, P.; Soldovieri, M.V.; Fontana, A.; Nardello, R.; Robbiano, A.; Bellini, G.; Elia, M.; Zara, F.; Taglialatela, M.; et al. A novel KCNQ3 mutation in familial epilepsy with focal seizures and intellectual disability. Epilepsia 2015, 56, e15–e20. [Google Scholar] [CrossRef] [PubMed]
- Shakespeare, W. The Tragedy of Hamlet, Prince of Denmark; The Folio Society: London, UK, 1623. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Present case | Ergun et al., 2004 [9] | Bonaglia et al., 2005 [10] | Wheeler, 2010 [11] | Concolino et al., 2012 [2] | Hilger et al., 2013 [12] | Rezek et al., 2014 [3] | Farcas et al., 2019 [13] | Lin et al., 2021 [14] | Sun et al., 2021 [15] | Total of cases with phenotype | ||||
| Index case | Mother | Case 1 | Case 2 | |||||||||||
| q24.13-q24.3 | q24.13-q24.3 | q22.3~q23-qter. | 8q24.3 | 8q23.3-q24.1 | 8q22.2-q24.2 | 8q24.3 | q23.3–q24.1 | q24.12-q24.3 | q24.12q24.3 | q24.12q24.3 | q24.3 | |||
| invdup(8) | invdup(8) | t(1;8) | invdup(8) | dup(8) | dup(8) | dup(8) | t(8;22) | dup del(8) | dup (8) x6~7 | dup (8) x6~7 | t(8;21) | |||
| GRCh38/hg38 | GRCh38/hg38 | NA | NA | NA | NCBI36/hg18 | NCBI36/hg18 | GRCh37/hg19 | GRCh37/hg19 | GRCh37/hg19 | GRCh37/hg19 | NA | |||
| 125385074–142496610 | 125385074–142496610 | NA | NA | 116147673–129419458 | 100338614–145464363 | 145012210–145132100 | 116147673–129419458 | 119488147–145984903 | 119328435–146295771 | 119261902–146295771 | 140238896–14629577 | |||
| Inheritance | Maternal | Unknown | de novo | de novo | Unknown | de novo | de novo | Maternal # | de novo | de novo | de novo | Paternal ## | ||
| Growth delay | - | - | + | + | - | + | NA | NA | NA | NA | NA | + | 4 | |
| Low weight | - | - | + | + | - | + | NA | NA | NA | NA | NA | + | 4 | |
| Short stature | - | - | - | + | - | + | NA | NA | NA | NA | NA | + | 3 | |
| Head and neck | Microcephaly | + | - | + | - | + | - | NA | NA | + | NA | NA | - | 4 |
| Prominent fronthead | - | - | + | NA | NA | + | NA | + | + | NA | NA | - | 4 | |
| Facial features | Hypertelorism | - | - | + | NA | + | + | NA | + | + | NA | NA | + | 6 |
| Palpebral slant alteration | - | Down | Up | NA | Down | NA | NA | NA | NA | NA | NA | Down | 4 | |
| Philtrum alterations | - | - | NA | Short | Short | Long | NA | NA | NA | NA | NA | Long or short * | 4 | |
| Microretrognathia | - | - | NA | NA | + | + | NA | NA | - | NA | NA | + | 3 | |
| Cleft lip/palate | - | + | - | NA | NA | NA | NA | + | + | NA | NA | - | 3 | |
| High palate | - | - | NA | + | NA | + | NA | NA | + | NA | NA | - | 3 | |
| Nasal bridge alteration | - | - | Depressed | NA | NA | Large | NA | Shallow | Broad | NA | NA | Broad | 5 | |
| Large nose | - | - | NA | + | NA | NA | NA | + | NA | NA | NA | + | 3 | |
| Low-set ears | - | - | + | NA | + | + | NA | + | + | NA | NA | - | 5 | |
| Large ears | - | - | NA | + | NA | NA | NA | NA | NA | NA | NA | + | 2 | |
| Protruding ears | - | - | NA | NA | + | - | NA | NA | - | NA | NA | + | 2 | |
| Visual anomalies | - | - | NA | NA | + | + | NA | NA | - | NA | NA | + | 3 | |
| Limb anomalies | - | - | NA | + | + | + | NA | NA | + | NA | NA | - | 4 | |
| Skin anomalies | Hypertrichosis | + | - | NA | NA | + | - | NA | NA | NA | NA | NA | NA | 2 |
| Neurologic aspects | Hypotonia | - | - | NA | NA | + | - | NA | NA | - | NA | NA | - | 1 |
| Seizures/epilepsy | + | - | NA | + | NA | - | NA | + | NA | NA | NA | - | 3 | |
| Learning disability | + | + | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | 2 | |
| Intelectual deficiency | + | - | + | + | + | + | NA | + | Unknown | NA | NA | + | 7 | |
| Motor delay | + | - | + | + | - | - | NA | + | NA | NA | NA | - | 4 | |
| Hearing loss | - | - | NA | NA | + | - | NA | NA | - | NA | NA | - | 1 | |
| Other systems | Heart defects | - | - | + | NA | + | + | + | NA | + | + | + | - | 7 |
| Skeletal abnormalities | - | - | - | Malar hypoplasia | NA | Flat occiput | Butterfly vertebra | NA | NA | NA | NA | Scoliosis | 4 | |
| Cryptorchidism | - | female | NA | female | female | + | + | female | female | NA | NA | - | 2 | |
| Anal imperforation | - | - | NA | NA | NA | NA | + | NA | + | NA | NA | + | 3 | |
| Hernia | - | - | Inguinal | NA | NA | Umbilical | NA | NA | NA | NA | NA | NA | 2 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mergener, R.; Nunes, M.R.; Böttcher, A.K.; Siqueira, M.B.; Peruzzo, H.F.; Merola, M.C.; Riegel, M.; Zen, P.R.G. invdup(8)(8q24.13q24.3)—A Complex Alteration and Its Clinical Consequences. Genes 2024, 15, 910. https://doi.org/10.3390/genes15070910
Mergener R, Nunes MR, Böttcher AK, Siqueira MB, Peruzzo HF, Merola MC, Riegel M, Zen PRG. invdup(8)(8q24.13q24.3)—A Complex Alteration and Its Clinical Consequences. Genes. 2024; 15(7):910. https://doi.org/10.3390/genes15070910
Chicago/Turabian StyleMergener, Rafaella, Marcela Rodrigues Nunes, Ana Kalise Böttcher, Monique Banik Siqueira, Helena Froener Peruzzo, Milene Carvalho Merola, Mariluce Riegel, and Paulo Ricardo Gazzola Zen. 2024. "invdup(8)(8q24.13q24.3)—A Complex Alteration and Its Clinical Consequences" Genes 15, no. 7: 910. https://doi.org/10.3390/genes15070910
APA StyleMergener, R., Nunes, M. R., Böttcher, A. K., Siqueira, M. B., Peruzzo, H. F., Merola, M. C., Riegel, M., & Zen, P. R. G. (2024). invdup(8)(8q24.13q24.3)—A Complex Alteration and Its Clinical Consequences. Genes, 15(7), 910. https://doi.org/10.3390/genes15070910