
Quantitative Trait Loci Mapping and Comparative Transcriptome Analysis of Fruit Weight (FW) in Watermelon (Citrullus lanatus L.)
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. High-Throughput Sequencing and Data Processing and Analysis
2.3. SNP/InDel Identification and Annotation
2.4. Construction of Genetic Map and Mapping QTL
2.5. BSA and Mapping Strategy
2.6. Transcriptome Analysis and Candidate Gene Prediction
3. Results
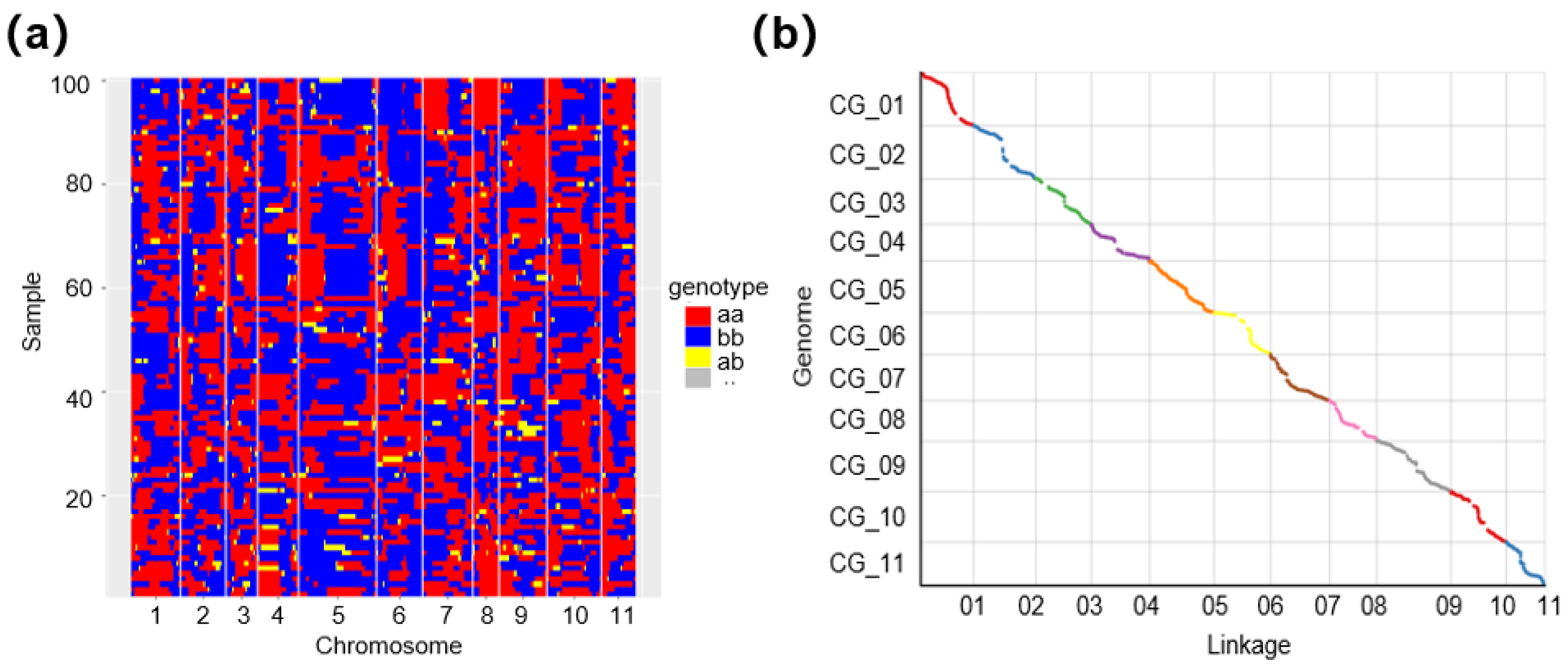
3.1. Population Establishment and Genetic Map Construction
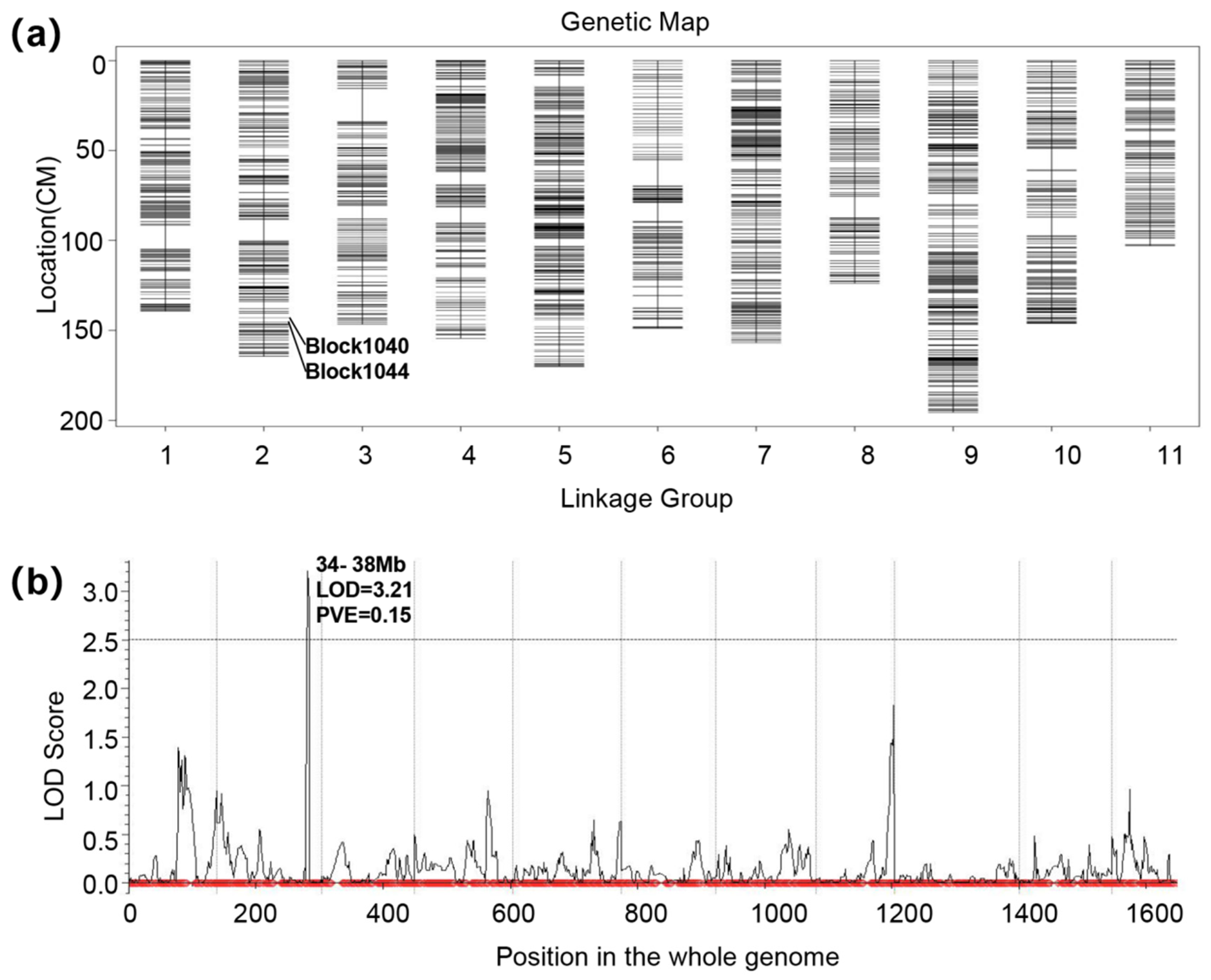
3.2. Evaluating the Genetic Map and Mapping QTL for FW
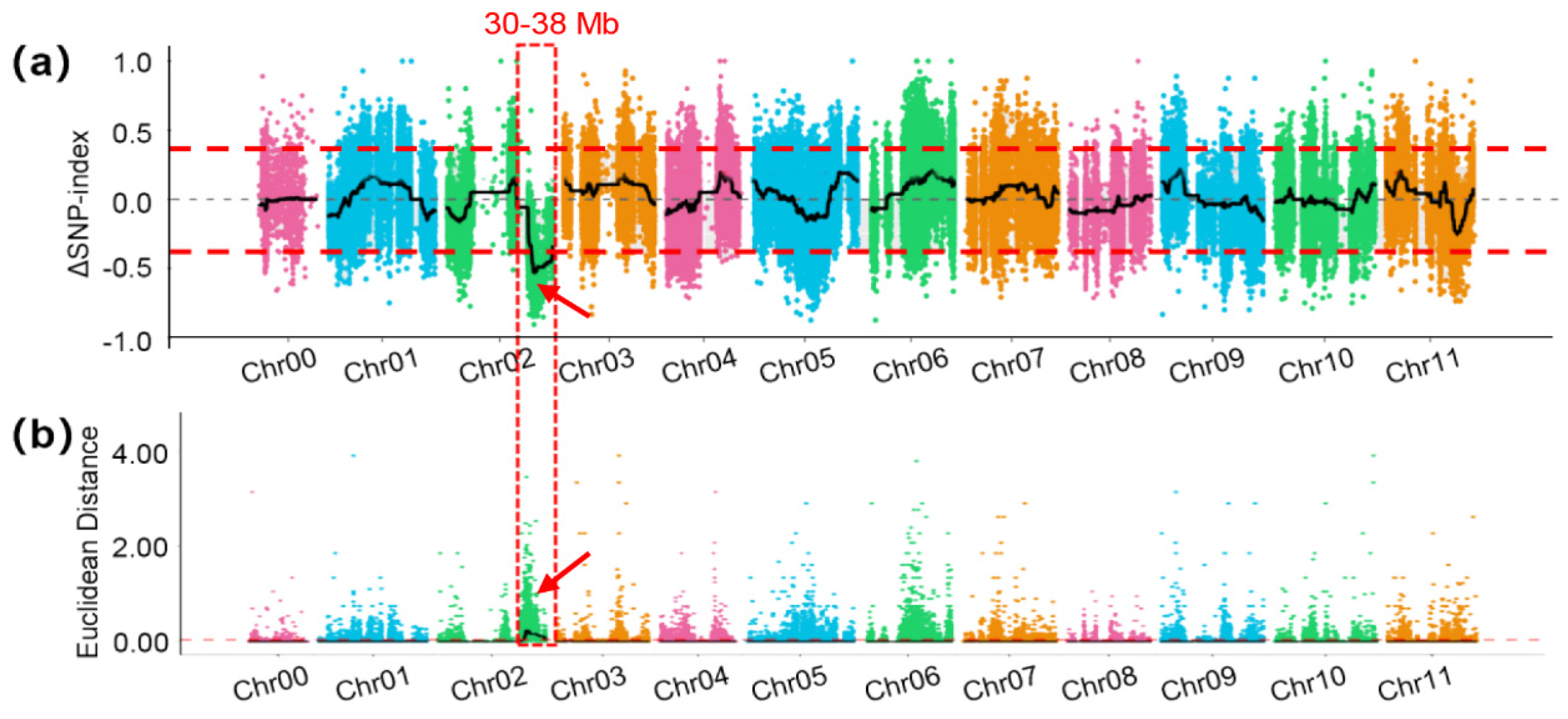
3.3. Bulked Segregant Analysis for FW
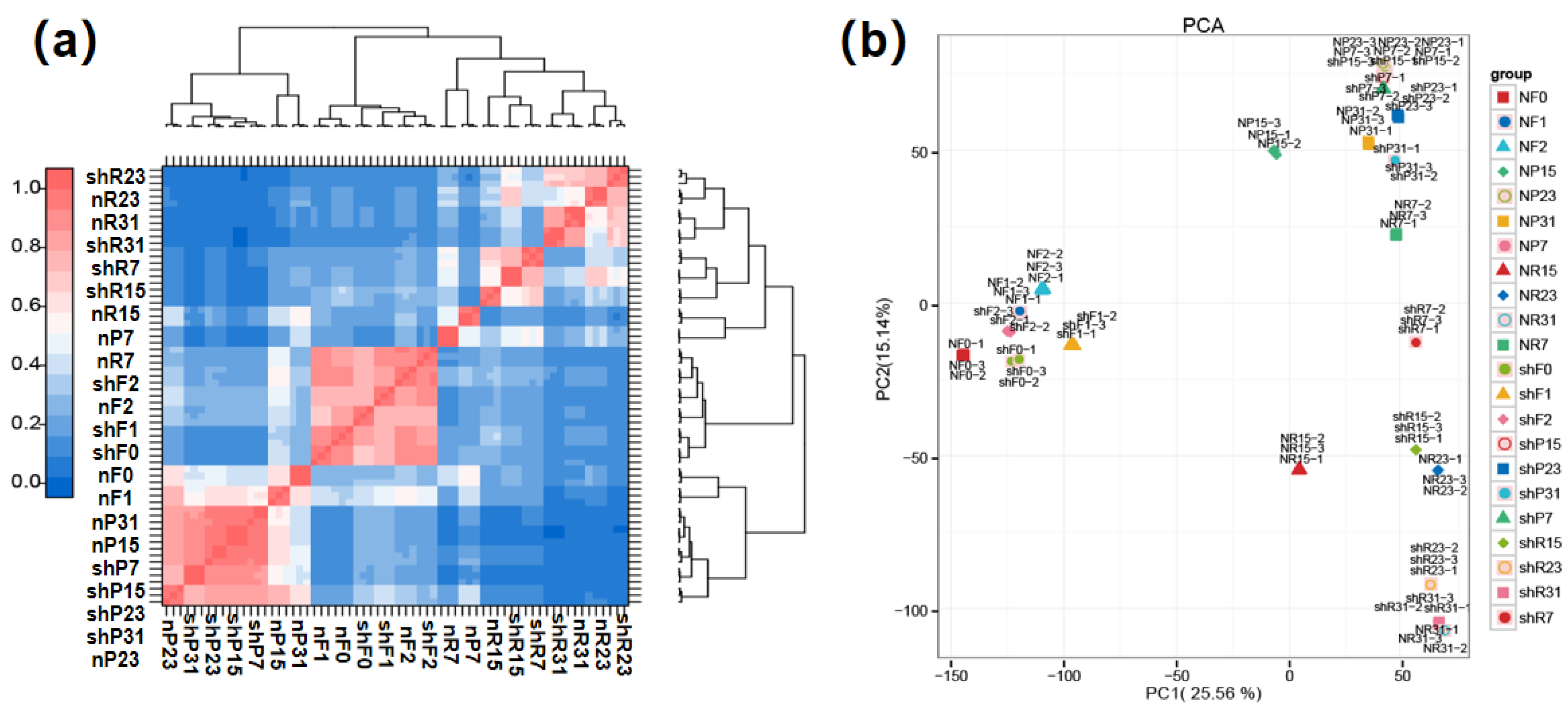
3.4. Multi-Tissue Gene Expression Profiles of Two Parental Lines
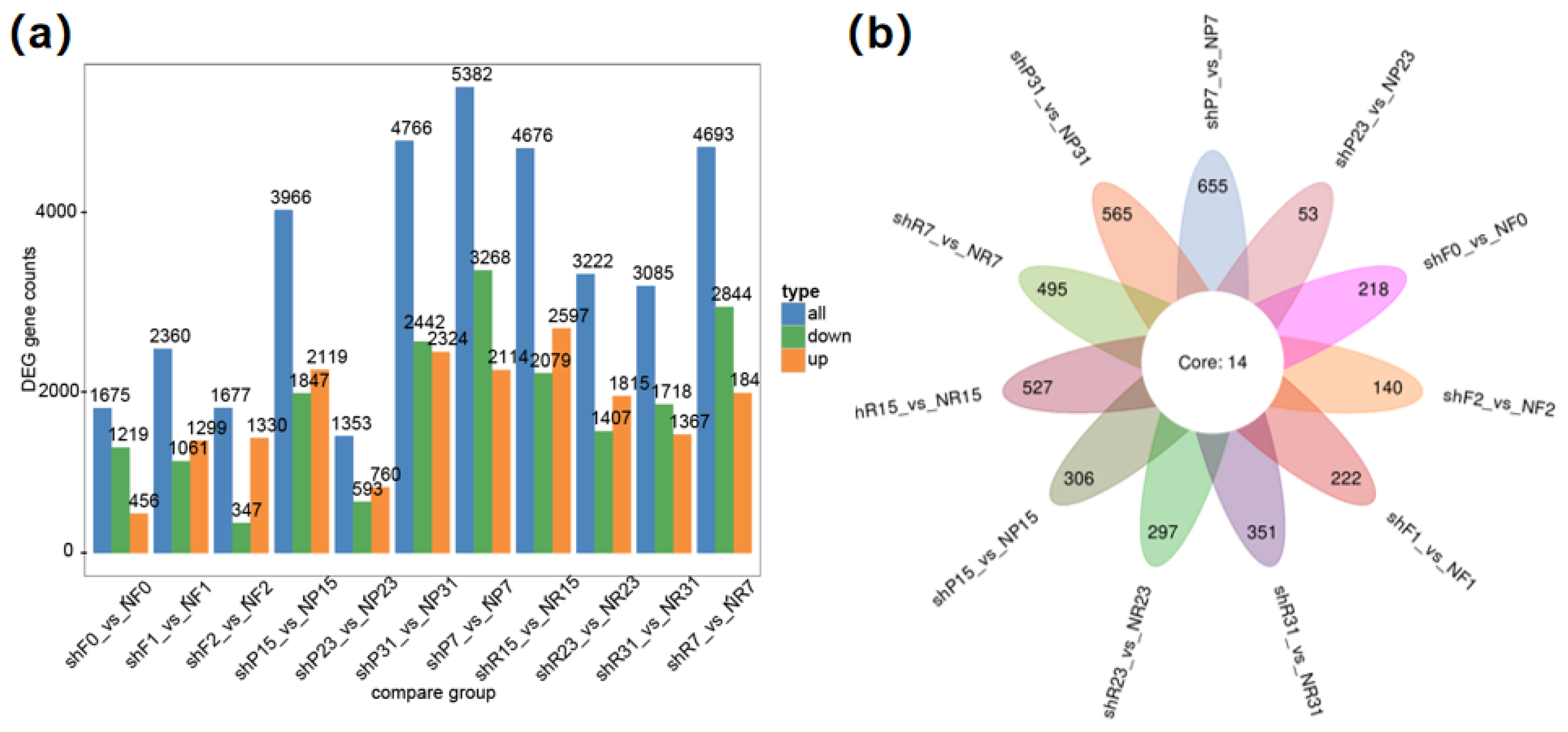
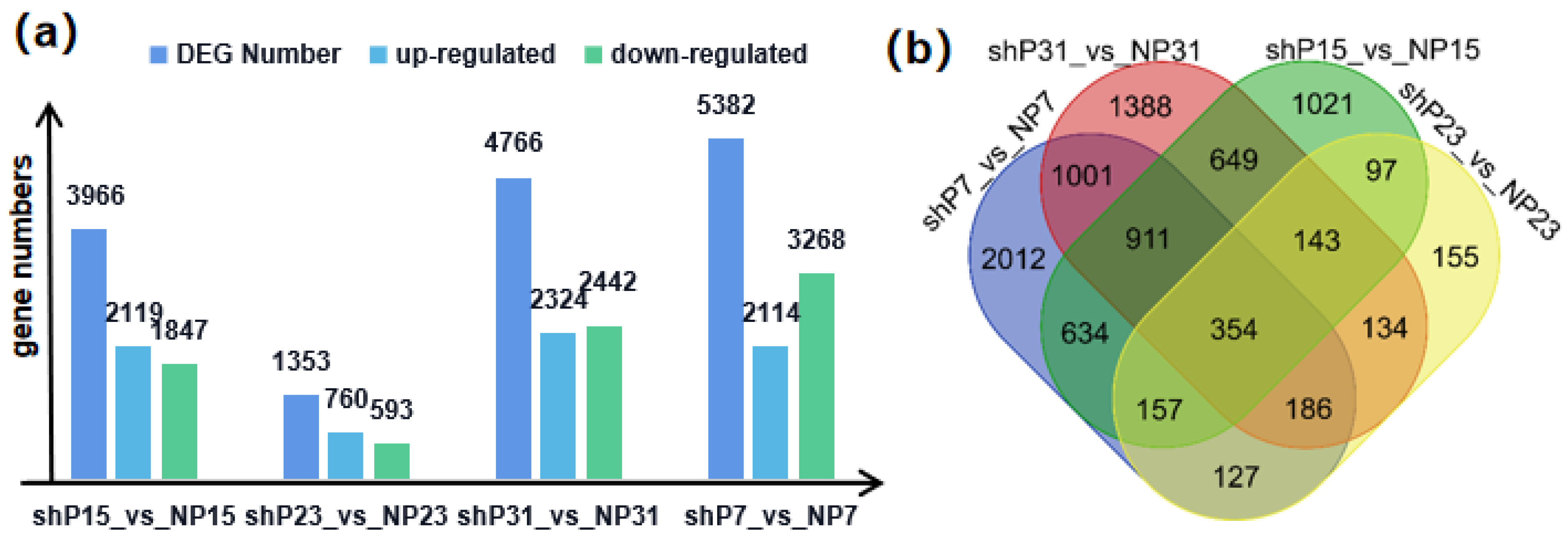
3.5. Identification of Differentially Expressed Genes (DEGs) Related to FW
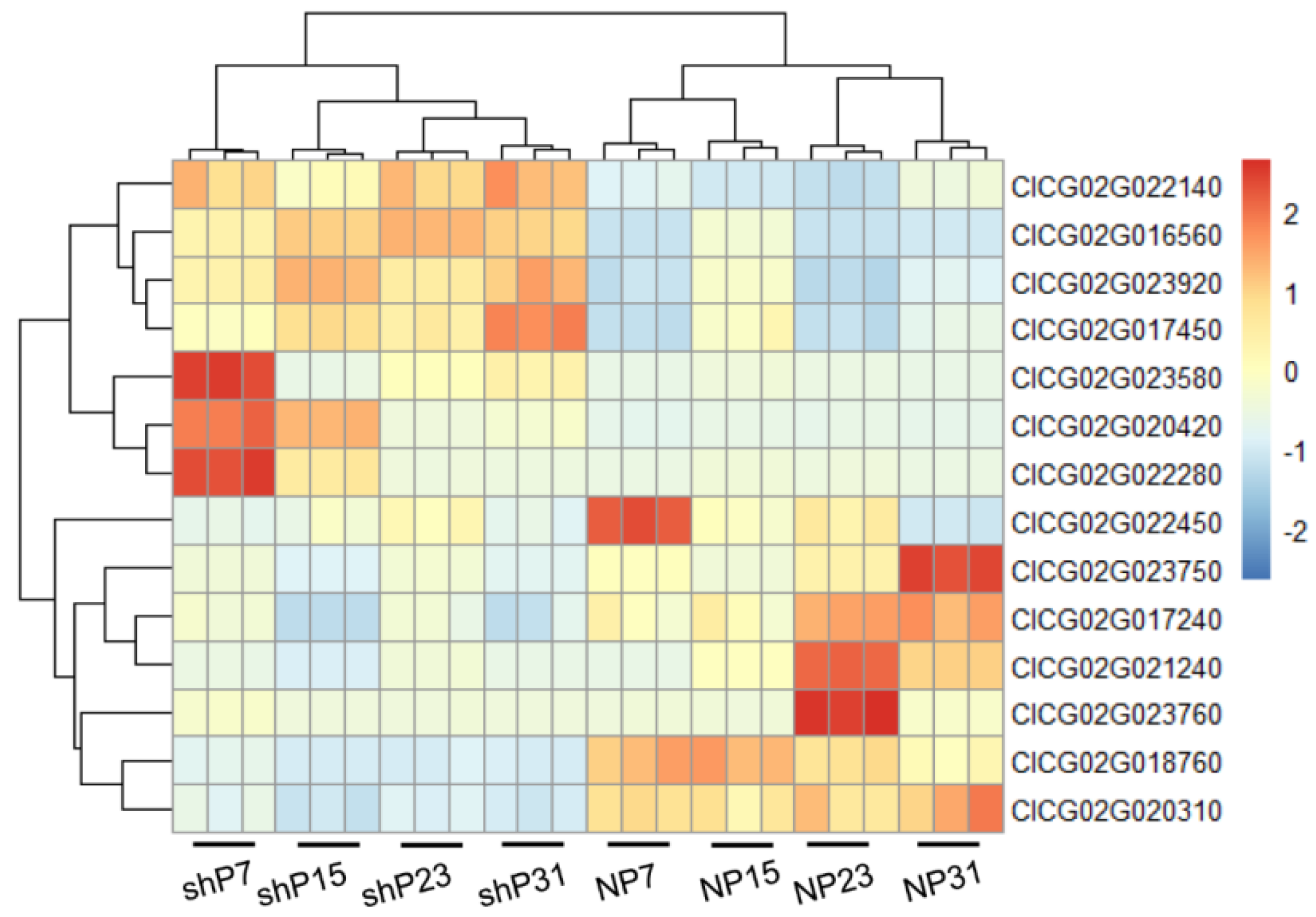
3.6. Co-Localization of QTL Mapping and Differentially Expressed Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AFLP | amplified fragment length polymorphisms |
| BSA | bulked segregant analysis |
| CIM | composite interval mapping |
| DEGs | differentially expressed genes |
| ED | Euclidean distance |
| FD | fruit width |
| FL | fruit length |
| FS | fruit shape |
| FSI | fruit shape indices |
| FW | fruit weight |
| LGs | linkage groups |
| NGS | next-generation sequencing |
| PCA | principal component analysis |
| PVE | phenotypic variation effect |
| QTL | quantitative trait loci |
| RILs | recombinant inbred lines |
| SNPs | single nucleotide polymorphisms |
| SSR | simple sequence repeat |
| WGR | whole-genome resequencing |
References
- Maoto, M.M.; Beswa, D.; Jideani, A.I.O. Watermelon as a potential fruit snack. Int. J. Food Prop. 2019, 22, 355–370. [Google Scholar] [CrossRef]
- Navot, N.; Zamir, D. Isozyme and seed protein phylogeny of the genusCitrullus (Cucurbitaceae). Plant Syst. Evol. 1987, 156, 61–67. [Google Scholar] [CrossRef]
- Zamir, D.; Navot, N.; Rudich, J. Enzyme polymorphism in Citrullus lanatus and C. colocynthis in Israel and Sinai. Plant Syst. Evol. 1984, 146, 163–170. [Google Scholar] [CrossRef]
- Hawkins, L.; Dane, F.; Kubisiak, T. Molecular Markers Associated with Morphological Traits in Watermelon. Am. Soc. Hortic. Sci. 2001, 36, 1318–1322. [Google Scholar] [CrossRef]
- Levi, A.; Thomas, C.; Trebitsh, T.; Salman, A.; King, J.; Karalius, J.; Newman, M.; Reddy, O.; Xu, Y.; Zhang, X. An extended linkage map for watermelon based on SRAP, AFLP, SSR, ISSR, and RAPD markers. J. Am. Soc. Hortic. Sci. 2006, 131, 393–402. [Google Scholar] [CrossRef]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, K.; Ossowski, S.; Lanz, C.; Juul, T.; Petersen, A.H.; Nielsen, K.L.; Jørgensen, J.-E.; Weigel, D.; Andersen, S.U. SHOREmap: Simultaneous mapping and mutation identification by deep sequencing. Nat. Methods. 2009, 6, 550–551. [Google Scholar] [CrossRef]
- Liu, S.; Yeh, C.-T.; Tang, H.M.; Nettleton, D.; Schnable, P.S. Gene Mapping via Bulked Segregant RNA-Seq (BSR-Seq). PLoS ONE 2012, 7, e36406. [Google Scholar] [CrossRef] [PubMed]
- Magwene, P.M.; Willis, J.H.; Kelly, J.K. The Statistics of Bulk Segregant Analysis Using Next Generation Sequencing. PLoS Comput. Biol. 2011, 7, e1002255. [Google Scholar] [CrossRef]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef]
- Hill, J.T.; Demarest, B.L.; Bisgrove, B.W.; Gorsi, B.; Su, Y.-C.; Yost, H.J. MMAPPR: Mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 2013, 23, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, X.; Pan, Q.; Li, P.; Liu, Y.; Lu, X.; Zhong, W.; Li, M.; Han, L.; Li, J.; et al. QTG-Seq Accelerates QTL Fine Mapping through QTL Partitioning and Whole-Genome Sequencing of Bulked Segregant Samples. Mol. Plant. 2019, 12, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, X.; Shi, S.; Zhang, H.; Wang, X.; Chen, H.; Li, W.; Li, L. DeepBSA: A deep-learning algorithm improves bulked segregant analysis for dissecting complex traits. Mol. Plant. 2022, 15, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zhang, J.; Sun, H.; Salse, J.; Lucas, W.J.; Zhang, H.; Zheng, Y.; Mao, L.; Ren, Y.; Wang, Z.; et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 2013, 45, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zhao, S.; Sun, H.; Wang, X.; Wu, S.; Lin, T.; Ren, Y.; Gao, L.; Deng, Y.; Zhang, J.; et al. Resequencing of 414 cultivated and wild watermelon accessions identifies selection for fruit quality traits. Nat. Genet. 2019, 51, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Renner, S.S.; Wu, S.; Pérez-Escobar, O.A.; Silber, M.V.; Fei, Z.; Chomicki, G. A chromosome-level genome of a Kordofan melon illuminates the origin of domesticated watermelons. Proc. Natl. Acad. Sci. USA 2021, 118, e2101486118. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wang, X.; Reddy, U.; Sun, H.; Bao, K.; Gao, L.; Mao, L.; Patel, T.; Ortiz, C.; Abburi, V.L.; et al. Genome of ‘Charleston Gray’, the principal American watermelon cultivar, and genetic characterization of 1,365 accessions in the U.S. National Plant Germplasm System watermelon collection. Plant Biotechnol. J. 2019, 17, 2246–2258. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Liu, S.; Zhang, Y.; Tan, J.; Li, X.; Chu, X.; Xu, B.; Tian, Y.; Sun, Y.; Li, B.; et al. A telomere-to-telomere gap-free reference genome of watermelon and its mutation library provide important resources for gene discovery and breeding. Mol. Plant. 2022, 15, 1268–1284. [Google Scholar] [CrossRef]
- Liu, S.; Gao, P.; Zhu, Q.; Luan, F.; Davis, A.R.; Wang, X. Development of cleaved amplified polymorphic sequence markers and a CAPS-based genetic linkage map in watermelon (Citrullus lanatus Matsum and Nakai) constructed using whole-genome re-sequencing data. Breed. Sci. 2016, 66, 244–259. [Google Scholar] [CrossRef]
- Ren, R.; Ray, R.; Li, P.; Xu, J.; Zhang, M.; Liu, G.; Yao, X.; Kilian, A.; Yang, X. Construction of a high-density DArTseq SNP-based genetic map and identification of genomic regions with segregation distortion in a genetic population derived from a cross between feral and cultivated-type watermelon. Mol. Genet. Genom. 2015, 290, 1457–1470. [Google Scholar] [CrossRef]
- Shang, J.; Li, N.; Li, N.; Xu, Y.; Ma, S.; Wang, J. Construction of a high-density genetic map for watermelon (Citrullus lanatus L.) based on large-scale SNP discovery by specific length amplified fragment sequencing (SLAF-seq). Sci. Hortic. 2016, 203, 38–46. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, Y.; McGregor, C.; Liu, S.; Luan, F.; Gao, M.; Weng, Y. Genetic architecture of fruit weight and shape variation in cucurbits: A comparative perspective. Theor. Appl. Genet. 2020, 133, 1–21. [Google Scholar]
- Wei, Q.Z.; Fu, W.Y.; Wang, Y.Z.; Qin, X.D.; Wang, J.; Li, J.; Lou, Q.F.; Chen, J.F. Rapid identification of fruit length loci in cucumber (Cucumis sativus L.) using next-generation sequencing (NGS)based QTL analysis. Sci. Rep. UK 2016, 6, 27496. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.P.; Liang, X.J.; Gao, M.L.; Liu, H.Q.; Meng, H.W.; Weng, Y.Q.; Cheng, Z.H. Round fruit shape in WI7239 cucumber is controlled by two interacting quantitative trait loci with one putatively encoding a tomato SUN homolog. Theor. Appl. Genet. 2017, 130, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.H.A.N.; Zhang, B.Y.; Keyhaninejad, N.; Rodríguez, G.R.; Kim, H.J.; Chakrabarti, M.; Illa-Berenguer, E.; Taitano, N.K.; Gonzalo, M.J.; Díaz, A. Tomato reveals a common mechanism underlying morphological diversification in domesticated plants. Nat. Commun. 2018, 9, 4734. [Google Scholar] [CrossRef] [PubMed]
- Sandlin, K.; Prothro, J.; Heesacker, A.; Khalilian, N.; Okashah, R.; Xiang, W.; Bachlava, E.; Caldwell, D.G.; Taylor, C.A.; Seymour, D.K.; et al. Comparative mapping in watermelon [Citrullus lanatus (Thunb.) Matsum. et Nakai]. Theor. Appl. Genet. 2012, 125, 1603–1618. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; McGregor, C.; Zhang, Y.; Gong, G.; Zhang, H.; Guo, S.; Sun, H.; Cai, W.; Zhang, J.; Xu, Y. An integrated genetic map based on four mapping populations and quantitative trait loci associated with economically important traits in watermelon (Citrullus lanatus). BMC Plant Biol. 2014, 14, 33. [Google Scholar]
- Kim, K.-H.; Hwang, J.-H.; Han, D.-Y.; Park, M.; Kim, S.; Choi, D.; Kim, Y.; Lee, G.P.; Kim, S.-T.; Park, Y.-H. Major Quantitative Trait Loci and Putative Candidate Genes for Powdery Mildew Resistance and Fruit-Related Traits Revealed by an Intraspecific Genetic Map for Watermelon (Citrullus lanatus var. lanatus). PLoS ONE 2015, 10, e0145665. [Google Scholar] [CrossRef] [PubMed]
- Dou, J.; Zhao, S.; Lu, X.; He, N.; Zhang, L.; Ali, A.; Kuang, H.; Liu, W. Genetic mapping reveals a candidate gene (ClFS1) for fruit shape in watermelon (Citrullus lanatus L.). Theor. Appl. Genet. 2018, 131, 947–958. [Google Scholar] [CrossRef]
- Tanaka, T.; Wimol, S.; Mizutani, T. Inheritance of fruit shape and seed size of watermelon. J. Jpn. Soc. Hortic. Sci. 1995, 64, 543–548. [Google Scholar] [CrossRef]
- Cheng, Y.; Luan, F.; Wang, X.; Gao, P.; Zhu, Z.; Liu, S.; Baloch, A.M.; Zhang, Y. Construction of a genetic linkage map of watermelon (Citrullus lanatus) using CAPS and SSR markers and QTL analysis for fruit quality traits. Sci. Hortic. 2016, 202, 25–31. [Google Scholar] [CrossRef]
- Ariyadasa, R.; Mascher, M.; Nussbaumer, T.; Schulte, D.; Frenkel, Z.; Poursarebani, N.; Zhou, R.; Steuernagel, B.; Gundlach, H.; Taudien, S.; et al. A sequence-ready physical map of barley anchored genetically by two million single-nucleotide polymorphisms. Plant Physiol. 2014, 164, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Trick, M.; Long, Y.; Meng, J.; Bancroft, I. Single nucleotide polymorphism (SNP) discovery in the polyploid Brassica napus using Solexa transcriptome sequencing. Plant Biotechnol. J. 2009, 7, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, A.; Huang, X.; Zhao, Q.; Dong, G.; Qian, Q.; Sang, T.; Han, B. Mapping 49 quantitative trait loci at high resolution through sequencing-based genotyping of rice recombinant inbred lines. Theor. Appl. Genet. 2011, 122, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Su, T.; Zhi, S.; Zhang, F.; Wang, W.; Zhang, D.; Zhao, X.; Yu, Y. Construction of a sequence-based bin map and mapping of QTLs for downy mildew resistance at four developmental stages in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Mol. Breed. 2016, 36, 44. [Google Scholar] [CrossRef]
- Zhang, D.; Li, H.; Wang, J.; Zhang, H.; Hu, Z.; Chu, S.; Lv, H.; Yu, D. High-Density Genetic Mapping Identifies New Major Loci for Tolerance to Low-Phosphorus Stress in Soybean. Front. Plant Sci. 2016, 7, 372. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lu, X.; Dou, J.; Aslam, A.; Gao, L.; Zhao, S.; He, N.; Liu, W. Construction of A High-Density Genetic Map and Mapping of Fruit Traits in Watermelon (Citrullus lanatus L.) Based on Whole-Genome Resequencing. Int. J. Mol. Sci. 2018, 19, 3268. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, Y.; Cui, H.; Zhang, L.; Sha, T.; Wang, C.; Fan, C.; Luan, F.; Wang, X. Linkage Mapping and Comparative Transcriptome Analysis of Firmness in Watermelon (Citrullus lanatus). Front. Plant Sci. 2020, 11, 831. [Google Scholar] [CrossRef]
- Li, J.; Han, R.; Li, R.; Xu, Q.; Li, M.; Tang, Y.; Li, J.; Wang, X.; Li, Z.; Li, Q.; et al. iBP-seq: An efficient and low-cost multiplex targeted genotyping and epigenotyping system. Crop J. 2023, 11, 1605–1610. [Google Scholar] [CrossRef]
- Shimomura, K.; Fukino, N.; Sugiyama, M.; Kawazu, Y.; Sakata, Y.; Yoshioka, Y. Quantitative trait locus analysis of cucumber fruit morphological traits based on image analysis. Euphytica 2017, 213, 138. [Google Scholar] [CrossRef]
- Xiao, H.; Jiang, N.; Schaffner, E.; Stockinger, E.J.; van der Knaap, E. A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science 2008, 319, 1527–1530. [Google Scholar] [CrossRef] [PubMed]
- Munos, S.; Ranc, N.; Botton, E.; Bérard, A.; Rolland, S.; Duffé, P.; Carretero, Y.; Le Paslier, M.C.; Delalande, C.; Bouzayen, M.; et al. Increase in Tomato Locule Number Is Controlled by Two Single-Nucleotide Polymorphisms Located Near WUSCHEL. Plant Physiol. 2011, 156, 2244–2254. [Google Scholar] [CrossRef] [PubMed]
- Cong, B.; Barrero, L.S.; Tanksley, S.D. Regulatory change in YABBY-like transcription factor led to evolution of extreme fruit size during tomato domestication. Nat. Genet. 2008, 40, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Liu, X. The Role of Auxin Amide Hydrolase SlILR5 in Tomato Fruit Expansion. Master’s Thesis, Shenyang Agricultural University, Shenyang, China, 2020. [Google Scholar]
- Lemaire-Chamley, M.; Koutouan, C.; Jorly, J.; Assali, J.; Yoshida, T.; Nogueira, M.; Tohge, T.; Ferrand, C.; Peres, L.E.; Asamizu, E.; et al. A chimeric TGA repressor slows down fruit maturation and ripening in tomato. Plant Cell Physiol. 2022, 63, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Chen, J.; Xie, Z.; Gao, J.; Ren, G.; Gao, S.; Zhou, X.; Kuai, B. Jasmonic acid promotes degreening via MYC2/3/4- and ANAC019/055/072-mediated regulation of major chlorophyll catabolic genes. Plant J. 2015, 84, 597–610. [Google Scholar] [CrossRef]
- Wang, W.-Q.; Wang, J.; Wu, Y.-Y.; Li, D.-W.; Allan, A.C.; Yin, X.-R. Genome-wide analysis of coding and non-coding RNA reveals a conserved miR164-NAC regulatory pathway for fruit ripening. New Phytol. 2020, 225, 1618–1634. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LG ID | Total Bin Marker | Total Distance (cM) | Average Distance (cM) | Max Gap (cM) | Gaps < 5 cM (%) |
|---|---|---|---|---|---|
| LG01 | 255 | 139.4 | 0.55 | 13.44 | 99.21% |
| LG02 | 234 | 164.39 | 0.71 | 11.95 | 99.57% |
| LG03 | 163 | 146.55 | 0.9 | 18.35 | 98.77% |
| LG04 | 211 | 154.58 | 0.74 | 8.92 | 96.19% |
| LG05 | 402 | 170.18 | 0.42 | 6.49 | 99.75% |
| LG06 | 236 | 148.78 | 0.63 | 14.5 | 98.72% |
| LG07 | 259 | 156.88 | 0.61 | 4.56 | 100% |
| LG08 | 135 | 123.92 | 0.92 | 11.95 | 99.25% |
| LG09 | 247 | 195.71 | 0.8 | 6.31 | 99.59% |
| LG10 | 280 | 146.09 | 0.52 | 11.95 | 98.92% |
| LG11 | 175 | 102.93 | 0.59 | 5.58 | 99.43% |
| Total | 2597 | 1649.42 | 0.64 | 18.35 | 96.19% |
| Gene ID | Chr | Start | End | Annotation |
|---|---|---|---|---|
| ClCG02G022140 | 2 | 36,608,687 | 36,616,809 | transcription factor TGA9-like |
| ClCG02G023920 | 2 | 38,267,515 | 38,268,888 | protein YLS3-like isoform X2 |
| ClCG02G016560 | 2 | 31,036,756 | 31,040,967 | protein ECERIFERUM 26-like |
| ClCG02G017450 | 2 | 31,950,483 | 31,952,384 | probable protein phosphatase 2C 52 |
| ClCG02G023580 | 2 | 37,923,184 | 37,929,184 | linoleate 13S-lipoxygenase 2-1 |
| ClCG02G022280 | 2 | 36,720,766 | 36,721,559 | NAC domain-containing protein 90 |
| ClCG02G020420 | 2 | 34,918,750 | 34,919,220 | zinc finger protein ZAT12-like |
| ClCG02G023750 | 2 | 38,110,302 | 38,131,226 | peroxidase 2-like |
| ClCG02G022450 | 2 | 36,884,417 | 36,885,709 | protein FAF-like, chloroplastic |
| ClCG02G017240 | 2 | 31,779,639 | 31,782,992 | probable E3 ubiquitin-protein ligase RHY1A |
| ClCG02G018760 | 2 | 33,491,641 | 33,496,029 | NAC domain-containing protein 96 |
| ClCG02G020310 | 2 | 34,854,388 | 34,855,109 | Glyoxalase_2 domain-containing protein |
| ClCG02G021240 | 2 | 35,752,425 | 35,755,753 | Lysine_decarbox domain-containing protein |
| ClCG02G023760 | 2 | 38,142,602 | 38,144,446 | peroxidase 2-like |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, S.; Tian, M.; Du, H.; Liu, S.; Yu, R.; Shen, H. Quantitative Trait Loci Mapping and Comparative Transcriptome Analysis of Fruit Weight (FW) in Watermelon (Citrullus lanatus L.). Genes 2024, 15, 933. https://doi.org/10.3390/genes15070933
Guo S, Tian M, Du H, Liu S, Yu R, Shen H. Quantitative Trait Loci Mapping and Comparative Transcriptome Analysis of Fruit Weight (FW) in Watermelon (Citrullus lanatus L.). Genes. 2024; 15(7):933. https://doi.org/10.3390/genes15070933
Chicago/Turabian StyleGuo, Song, Mei Tian, Huiying Du, Shengfeng Liu, Rong Yu, and Huolin Shen. 2024. "Quantitative Trait Loci Mapping and Comparative Transcriptome Analysis of Fruit Weight (FW) in Watermelon (Citrullus lanatus L.)" Genes 15, no. 7: 933. https://doi.org/10.3390/genes15070933