Interstitial Lung Diseases and Non-Small Cell Lung Cancer: Particularities in Pathogenesis and Expression of Driver Mutations

 , and
, and
Abstract
:1. Introduction
2. Classification of Interstitial Lung Disease
3. Idiopathic Pulmonary Fibrosis
3.1. Wnt/β Catenin Signaling Pathway
3.2. Notch Signaling Pathway
3.3. TGF-β Signaling Pathway
3.4. MUC5B
3.5. TOLLIP
3.6. Telomerase-Related Mutations (TRM)
4. ILD and NSCLC
5. Driver Mutations in Patients with NSCLC and ILD
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, T.; De Los Santos, F.G.; Phan, S.H. The Bleomycin Model of Pulmonary Fibrosis. Methods Mol. Biol. 2017, 1627, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, K.M.; Margaritopoulos, G.A.; Tomassetti, S.; Bonella, F.; Costabel, U.; Poletti, V. Interstitial Lung Disease. Eur. Respir. Rev. 2014, 23, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Perelas, A.; Silver, R.M.; Arrossi, A.V.; Highland, K.B. Systemic Sclerosis-Associated Interstitial Lung Disease. Lancet Respir. Med. 2020, 8, 304–320. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Tomassetti, S.; Wells, A.U.; Costabel, U.; Cavazza, A.; Colby, T.V.; Rossi, G.; Sverzellati, N.; Carloni, A.; Carretta, E.; Buccioli, M.; et al. Bronchoscopic Lung Cryobiopsy Increases Diagnostic Confidence in the Multidisciplinary Diagnosis of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Antoniou, K.M.; Bissell, B.D.; Bouros, D.; Buendia-Roldan, I.; Caro, F.; Crestani, B.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, E18–E47. [Google Scholar] [CrossRef]
- Graney, B.A.; Fischer, A. Interstitial Pneumonia with Autoimmune Features. Ann. Am. Thorac. Soc. 2019, 16, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, M.; Faverio, P.; Manfredi, A.; Cassone, G.; Vacchi, C.; Stainer, A.; Pozzi, M.R.; Salvarani, C.; Pesci, A.; Luppi, F. Biomedicines Interstitial Pneumonia with Autoimmune Features: Why Rheumatologist-Pulmonologist Collaboration Is Essential. Biomedicines 2020, 9, 17. [Google Scholar] [CrossRef]
- Copeland, C.R.; Lancaster, L.H. Management of Progressive Fibrosing Interstitial Lung Diseases (PF-ILD). Front. Med. 2021, 8, 743977. [Google Scholar] [CrossRef]
- George, P.M.; Spagnolo, P.; Kreuter, M.; Altinisik, G.; Bonifazi, M.; Martinez, F.J.; Molyneaux, P.L.; Renzoni, E.A.; Richeldi, L.; Tomassetti, S.; et al. Progressive Fibrosing Interstitial Lung Disease: Clinical Uncertainties, Consensus Recommendations, and Research Priorities. Lancet Respir. Med. 2020, 8, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Hatabu, H.; Hunninghake, G.M.; Richeldi, L.; Brown, K.K.; Wells, A.U.; Remy-Jardin, M.; Verschakelen, J.; Nicholson, A.G.; Beasley, M.B.; Christiani, D.C.; et al. Interstitial Lung Abnormalities Detected Incidentally on CT: A Position Paper from the Fleischner Society. Lancet Respir. Med. 2020, 8, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Mei, Q.; Liu, Z.; Zuo, H.; Yang, Z.; Qu, J. Idiopathic Pulmonary Fibrosis: An Update on Pathogenesis. Front. Pharmacol. 2022, 12, 797292. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Collard, H.R. Epidemiology of Idiopathic Pulmonary Fibrosis. Clin. Epidemiol. 2013, 5, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Bendstrup, E.; Dron, L.; Langley, J.; Smith, G.; Khalid, J.M.; Patel, H.; Kreuter, M. Global Incidence and Prevalence of Idiopathic Pulmonary Fibrosis. Respir. Res. 2021, 22, 197. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K. Update on Immunohistochemistry for the Diagnosis of Lung Cancer. Cancers 2018, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Moss, B.J.; Ryter, S.W.; Rosas, I.O. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. 2022, 17, 515–546. [Google Scholar] [CrossRef] [PubMed]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.-I.; Taylor, C.J.; Jetter, C.; et al. Single-Cell RNA Sequencing Reveals Profibrotic Roles of Distinct. Epithelial and Mesenchymal Lineages in Pulmonary Fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Park, J.E.; Tsagkogeorga, G.; Yanagita, M.; Koo, B.K.; Han, N.; Lee, J.H. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors That Mediate Alveolar Regeneration. Cell Stem Cell 2020, 27, 366–382.e7. [Google Scholar] [CrossRef]
- Weidenfeld, J.; Shu, W.; Zhang, L.; Millar, S.E.; Morrisey, E.E. The WNT7b Promoter Is Regulated by TTF-1, GATA6, and Foxa2 in Lung Epithelium. J. Biol. Chem. 2002, 277, 21061–21070. [Google Scholar] [CrossRef]
- Mucenski, M.L.; Nation, J.M.; Thitoff, A.R.; Besnard, V.; Xu, Y.; Wert, S.E.; Harada, N.; Taketo, M.M.; Stahlman, M.T.; Whitsett, J.A.; et al. Catenin Regulates Differentiation of Respiratory Epithelial Cells in Vivo. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef] [PubMed]
- Chilosi, M.; Poletti, V.; Zamò, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/-Catenin Pathway Activation in Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2003, 162, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-Cell Wnt Signaling Niches Maintain Stemness of Alveolar Type 2 Cells. Science (1979) 2018, 359, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Königshoff, M.; Balsara, N.; Pfaff, E.M.; Kramer, M.; Chrobak, I.; Seeger, W.; Eickelberg, O. Functional Wnt Signaling Is Increased in Idiopathic Pulmonary Fibrosis. PLoS ONE 2008, 3, e0002142. [Google Scholar] [CrossRef] [PubMed]
- Aumiller, V.; Balsara, N.; Wilhelm, J.; Günther, A.; Königshoff, M. WNT/β-Catenin Signaling Induces IL-1β Expression by Alveolar Epithelial Cells in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2013, 49, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Morali, O.G.; Ronique Delmas, V.; Moore, R.; Jeanney, C.; Thiery, J.P.; Larue, L. IGF-II Induces Rapid b-Catenin Relocation to the Nucleus during Epithelium to Mesenchyme Transition. Oncogene 2001, 20, 4942–4950. [Google Scholar] [CrossRef]
- Samarelli, A.V.; Tonelli, R.; Marchioni, A.; Bruzzi, G.; Gozzi, F.; Andrisani, D.; Castaniere, I.; Manicardi, L.; Moretti, A.; Tabbì, L.; et al. Fibrotic Idiopathic Interstitial Lung Disease: The Molecular and Cellular Key Players. Int. J. Mol. Sci. 2021, 22, 8952. [Google Scholar] [CrossRef]
- Chilosi, M.; Caliò, A.; Rossi, A.; Gilioli, E.; Pedica, F.; Montagna, L.; Pedron, S.; Confalonieri, M.; Doglioni, C.; Ziesche, R.; et al. Epithelial to Mesenchymal Transition-Related Proteins ZEB1, β-Catenin, and β-Tubulin-III in Idiopathic Pulmonary Fibrosis. Mod. Pathol. 2017, 30, 26–38. [Google Scholar] [CrossRef]
- Marmai, C.; Sutherland, R.E.; Kim, K.K.; Dolganov, G.M.; Fang, X.; Kim, S.S.; Jiang, S.; Golden, J.A.; Hoopes, C.W.; Matthay, M.A.; et al. Alveolar Epithelial Cells Express Mesenchymal Proteins in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2011, 301, L71–L78. [Google Scholar] [CrossRef]
- Hill, C.; Jones, M.G.; Davies, D.E.; Wang, Y. Epithelial-Mesenchymal Transition Contributes to Pulmonary Fibrosis via Aberrant Epithelial/Fibroblastic Cross-Talk Introduction and Discussion. J. Lung Health Dis. 2019, 3, 31–35. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, K.; Nieuwenhuis, E.; Cohen, B.L.; Wang, W.; Canty, A.J.; Danska, J.S.; Coultas, L.; Rossant, J.; Wu, M.Y.; Piscione, T.D.; et al. Lunatic Fringe-Mediated Notch Signaling Is Required for Lung Alveogenesis. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 298, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Liu, J.; Wu, Z.; Liu, T.; Ullenbruch, M.R.; Ding, L.; Henke, C.A.; Bitterman, P.B.; Phan, S.H. Reemergence of Hedgehog Mediates Epithelial-Mesenchymal Crosstalk in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 52, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Froidure, A.; Marchal-Duval, E.; Homps-Legrand, M.; Ghanem, M.; Justet, A.; Crestani, B.; Mailleux, A. Chaotic Activation of Developmental Signalling Pathways Drives Idiopathic Pulmonary Fibrosis. Eur. Respir. Rev. 2020, 29, 190140. [Google Scholar] [CrossRef] [PubMed]
- Broekelmann, T.J.; Limper, A.H.; Colbyt, T.V.; Mcdonald, J.A. Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 1991, 88, 6642–6646. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.E.; Eickelberg, O. The Impact of TGF-β on Lung Fibrosis: From Targeting to Biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Transforming Growth Factor–ß in Tissue Fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Ye, Z.; Hu, Y. TGF-Β1: Gentlemanly Orchestrator in Idiopathic Pulmonary Fibrosis (Review). Int. J. Mol. Med. 2021, 48, 132. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-Based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A Common MUC5B Promoter Polymorphism and Pulmonary Fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef]
- Platenburg, M.G.J.P.; Wiertz, I.A.; van der Vis, J.J.; Crestani, B.; Borie, R.; Dieude, P.; Kannengiesser, C.; Burgers, J.A.; Grutters, J.C.; van Moorsel, C.H.M. The MUC5B Promoter Risk Allele for Idiopathic Pulmonary Fibrosis Predisposes to Asbestosis. Eur. Respir. J. 2020, 55, 1902361. [Google Scholar] [CrossRef] [PubMed]
- Biondini, D.; Cocconcelli, E.; Bernardinello, N.; Lorenzoni, G.; Rigobello, C.; Lococo, S.; Castelli, G.; Baraldo, S.; Cosio, M.G.; Gregori, D.; et al. Prognostic Role of MUC5B Rs35705950 Genotype in Patients with Idiopathic Pulmonary Fibrosis (IPF) on Antifibrotic Treatment. Respir. Res. 2021, 22, 98. [Google Scholar] [CrossRef] [PubMed]
- Oldham, J.M.; Ma, S.F.; Martinez, F.J.; Anstrom, K.J.; Raghu, G.; Schwartz, D.A.; Valenzi, E.; Witt, L.; Lee, C.; Vij, R.; et al. TOLLIP, MUC5B, and the Response to N-Acetylcysteine among Individuals with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Goobie, G.C.; Gregory, A.D.; Kass, D.J.; Zhang, Y. Toll-Interacting Protein in Pulmonary Diseases Abiding by the Goldilocks Principle. Am. J. Respir. Cell Mol. Biol. 2021, 64, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Isshiki, T.; Koyama, K.; Homma, S.; Sakamoto, S.; Yamasaki, A.; Shimizu, H.; Miyoshi, S.; Nakamura, Y.; Kishi, K. Association of Rs3750920 Polymorphism in TOLLIP with Clinical Characteristics of Fibrosing Interstitial Lung Diseases in Japanese. Sci. Rep. 2021, 11, 16250. [Google Scholar] [CrossRef] [PubMed]
- Muraki, K.; Nyhan, K.; Han, L.; Murnane, J.P. Mechanisms of Telomere Loss and Their Consequences for Chromosome Instability. Front. Oncol. 2012, 2, 135. [Google Scholar] [CrossRef] [PubMed]
- Courtwright, A.M.; El-Chemaly, S. Telomeres in Interstitial Lung Disease: The Short and the Long of It. Ann. Am. Thorac. Soc. 2019, 16, 175–181. [Google Scholar] [CrossRef]
- Newton, C.A.; Batra, K.; Torrealba, J.; Kozlitina, J.; Glazer, C.S.; Aravena, C.; Meyer, K.; Raghu, G.; Collard, H.R.; Garcia, C.K. Telomere-Related Lung Fibrosis Is Diagnostically Heterogeneous but Uniformly Progressive. Eur. Respir. J. 2016, 48, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.I.; Park, S.H.; Park, B.J.; Lee, C.W. Interstitial Lung Disease and Lung Cancer Development: A 5-Year Nationwide Population-Based Study. Cancer Res. Treat. 2018, 50, 374–381. [Google Scholar] [CrossRef]
- JafariNezhad, A.R.; YektaKooshali, M.H. Lung Cancer in Idiopathic Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. PLoS ONE 2018, 13, e0202360. [Google Scholar] [CrossRef]
- Frank, A.J.; Dagogo-Jack, I.; Dobre, I.A.; Tait, S.; Schumacher, L.; Fintelmann, F.J.; Fingerman, L.M.; Keane, F.K.; Montesi, S.B. Management of Lung Cancer in the Patient with Interstitial Lung Disease. Oncologist 2023, 28, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Mattoo, H.; Pillai, S. Idiopathic Pulmonary Fibrosis and Systemic Sclerosis: Pathogenic Mechanisms and Therapeutic Interventions. Cell. Mol. Life Sci. 2021, 78, 5527–5542. [Google Scholar] [CrossRef]
- Karampitsakos, T.; Spagnolo, P.; Mogulkoc, N.; Wuyts, W.A.; Tomassetti, S.; Bendstrup, E.; Molina-Molina, M.; Manali, E.D.; Unat, Ö.S.; Bonella, F.; et al. Lung Cancer in Patients with Idiopathic Pulmonary Fibrosis: A Retrospective Multicentre Study in Europe. Respirology 2023, 28, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, Y.; Suda, T.; Naito, T.; Enomoto, N.; Hashimoto, D.; Fujisawa, T.; Nakamura, Y.; Inui, N.; Nakamura, H.; Chida, K. Cumulative Incidence of and Predictive Factors for Lung Cancer in IPF. Respirology 2009, 14, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Jeong, B.H.; Chung, M.J.; Lee, K.S.; Kwon, O.J.; Chung, M.P. Risk Factors and Clinical Characteristics of Lung Cancer in Idiopathic Pulmonary Fibrosis: A Retrospective Cohort Study. BMC Pulm. Med. 2019, 19, 149. [Google Scholar] [CrossRef]
- Tomassetti, S.; Gurioli, C.; Ryu, J.H.; Decker, P.A.; Ravaglia, C.; Tantalocco, P.; Buccioli, M.; Piciucchi, S.; Sverzellati, N.; Dubini, A.; et al. The Impact of Lung Cancer on Survival of Idiopathic Pulmonary Fibrosis. Chest 2015, 147, 157–164. [Google Scholar] [CrossRef]
- Hida, T.; Hata, A.; Lu, J.; Valtchinov, V.I.; Hino, T.; Nishino, M.; Honda, H.; Tomiyama, N.; Christiani, D.C.; Hatabu, H. Interstitial Lung Abnormalities in Patients with Stage I Non-Small Cell Lung Cancer Are Associated with Shorter Overall Survival: The Boston Lung Cancer Study. Cancer Imaging 2021, 21, 14. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.J.; Do, K.H.; Lee, J.B.; Alblushi, S.; Lee, S.M. Lung Cancer in Combined Pulmonary Fibrosis and Emphysema: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0161437. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Ogura, T.; Yokose, T.; Nagai, K.; Nishiwaki, Y.; Esumi, H. P53 Gene Alteration in Atypical Epithelial Lesions and Carcinoma in Patients with Idiopathic Pulmonary Fibrosis. Hum. Pathol. 2001, 32, 1043–1049. [Google Scholar] [CrossRef]
- Takahashi, T.; Munakata, M.; Ohtsuka, Y.; Nisihara, H.; Nasuhara, Y.; Kamachi-Satoh, A.; Dosaka-Akita, H.; Homma, Y.; Kawakami, Y. Expression and Alteration of Ras and P53 Proteins in Patients with Lung Carcinoma Accompanied by Idiopathic Pulmonary Fibrosis. Cancer 2002, 95, 624–633. [Google Scholar] [CrossRef]
- Ballester, B.; Milara, J.; Cortijo, J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2019, 20, 593. [Google Scholar] [CrossRef]
- Xie, B.; Zheng, G.; Li, H.; Yao, X.; Hong, R.; Li, R.; Yue, W.; Chen, Y. Effects of the Tumor Suppressor PTEN on the Pathogenesis of Idiopathic Pulmonary Fibrosis in Chinese Patients. Mol. Med. Rep. 2016, 13, 2715–2723. [Google Scholar] [CrossRef] [PubMed]
- Sirhan, Z.; Alojair, R.; Thyagarajan, A.; Sahu, R.P. Therapeutic Implications of PTEN in Non-Small Cell Lung Cancer. Pharmaceutics 2023, 15, 2090. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, M.; Li, N.; Da Yan, L.; Zhou, W.; Yu, Z.Q.; Peng, X.C.; Cai, J.; Yang, Y.H. TERT Mutations in Non–Small Cell Lung Cancer: Clinicopathologic Features and Prognostic Implications. Clin. Med. Insights Oncol. 2023, 17, 11795549221140781. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, X.; Long, Q.; Wang, X.; Zhu, W.; Chen, E.; Zhou, W.; Yang, H.; Huang, C.; Deng, W.; et al. TERC Promotes Non-Small Cell Lung Cancer Progression by Facilitating the Nuclear Localization of TERT. iScience 2024, 27. [Google Scholar] [CrossRef] [PubMed]
- Diaz de Leon, A.; Cronkhite, J.T.; Katzenstein, A.L.A.; Godwin, J.D.; Raghu, G.; Glazer, C.S.; Rosenblatt, R.L.; Girod, C.E.; Garrity, E.R.; Xing, C.; et al. Telomere Lengths, Pulmonary Fibrosis and Telomerase (TERT) Mutations. PLoS ONE 2010, 5, e0010680. [Google Scholar] [CrossRef] [PubMed]
- Borie, R.; Tabèze, L.; Thabut, G.; Nunes, H.; Cottin, V.; Marchand-Adam, S.; Prevot, G.; Tazi, A.; Cadranel, J.; Mal, H.; et al. Prevalence and Characteristics of TERT and TERC Mutations in Suspected Genetic Pulmonary Fibrosis. Eur. Respir. J. 2016, 48, 1721–1731. [Google Scholar] [CrossRef]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-Associated Fibroblasts: Overview, Progress, Challenges, and Directions. Cancer Gene Ther. 2021, 28, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Kim, H.H.; Hyun, D.G.; Ji, W.; Choi, C.M.; Lee, J.C.; Kim, H.C. Clinical Characteristics and Outcome of Lung Cancer in Patients with Fibrosing Interstitial Lung Disease. BMC Pulm. Med. 2024, 24, 136. [Google Scholar] [CrossRef]
- Terasaki, F.; Azuma, A.; Anzai, T.; Ishizaka, N.; Ishida, Y.; Isobe, M.; Inomata, T.; Ishibashi-Ueda, H.; Eishi, Y.; Kitakaze, M.; et al. JCS 2016 Guideline on Diagnosis and Treatment of Cardiac Sarcoidosis—Digest Version—. Circ. J. 2019, 83, 2329–2388. [Google Scholar] [CrossRef]
- Jankowich, M.D.; Rounds, S.I.S. Combined Pulmonary Fibrosis and Emphysema Syndrome: A Review. Chest 2012, 141, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Tzouvelekis, A.; Antoniou, K.; Kreuter, M.; Evison, M.; Blum, T.G.; Poletti, V.; Grigoriu, B.; Vancheri, C.; Spagnolo, P.; Karampitsakos, T.; et al. The Diamorfosis (Diagnosis and Management of Lung Cancer and Fibrosis) Survey: International Survey and Call for Consensus. ERJ Open Res. 2021, 7, 00529–02020. [Google Scholar] [CrossRef] [PubMed]
- Grodkiewicz, M.; Koziel, P.; Chmielewska, I.; Korbel, M.A.; Milanowski, J. Small Cell Lung Cancer in the Course of Idiopathic Pulmonary Fibrosis—Case Report and Literature Review. Curr. Oncol. 2022, 29, 5077–5083. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Shimizu, Y.; Goto, T.; Kitahara, A.; Koike, T.; Ishikawa, H.; Watanabe, T.; Tsuchida, M. Survival after Repeated Surgery for Lung Cancer with Idiopathic Pulmonary Fibrosis: A Retrospective Study. BMC Pulm. Med. 2018, 18, 134. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Kiura, K.; Takigawa, N.; Yoshioka, H.; Harita, S.; Kuyama, S.; Yonei, T.; Fujiwara, K.; Maeda, T.; Aoe, K.; et al. Comparison of the Incidence and Pattern of Interstitial Lung Disease during Erlotinib and Gefitinib Treatment in Japanese Patients with Non-Small Cell Lung Cancer: The Okayama Lung Cancer Study Group Experience. J. Thorac. Oncol. 2010, 5, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Kudoh, S.; Kato, H.; Nishiwaki, Y.; Fukuoka, M.; Nakata, K.; Ichinose, Y.; Tsuboi, M.; Yokota, S.; Nakagawa, K.; Suga, M.; et al. Interstitial Lung Disease in Japanese Patients with Lung Cancer: A Cohort and Nested Case-Control Study. Am. J. Respir. Crit. Care Med. 2008, 177, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Suh, C.H.; Kim, K.W.; Pyo, J.; Hatabu, H.; Nishino, M. The Incidence of ALK Inhibitor-Related Pneumonitis in Advanced Non-Small-Cell Lung Cancer Patients: A Systematic Review and Meta-Analysis. Lung Cancer 2019, 132, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Qie, W.; Zhao, Q.; Yang, L.; Zou, B.; Duan, Y.; Yao, Y.Y.; Wang, L. Incidence of Pneumonitis Following the Use of Different Anaplastic Lymphoma Kinase Tyrosine Kinase Inhibitor Regimens: An Updated Systematic Review and Meta-Analysis. Cancer Med. 2023, 12, 13873–13884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J. Cellular and Molecular Mechanisms in Idiopathic Pulmonary Fibrosis. Adv. Respir. Med. 2023, 91, 26–48. [Google Scholar] [CrossRef]
- Stella, G.M.; D’Agnano, V.; Piloni, D.; Saracino, L.; Lettieri, S.; Mariani, F.; Lancia, A.; Bortolotto, C.; Rinaldi, P.; Falanga, F.; et al. The Oncogenic Landscape of the Idiopathic Pulmonary Fibrosis: A Narrative Review. Transl. Lung Cancer Res. 2022, 11, 472–496. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Mishra, L. Role of TGF-β in Stem Cells and Cancer. Oncogene 2005, 24, 5667. [Google Scholar] [CrossRef]
- Hata, A.; Nakajima, T.; Matsusaka, K.; Fukuyo, M.; Nakayama, M.; Morimoto, J.; Ito, Y.; Yamamoto, T.; Sakairi, Y.; Rahmutulla, B.; et al. Genetic Alterations in Squamous Cell Lung Cancer Associated with Idiopathic Pulmonary Fibrosis. Int. J. Cancer 2021, 148, 3008–3018. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, Z.; Xuan, Y.; Zhao, Y.; Deng, C.; Wang, M.; Xie, C.; Yuan, F.; Pang, Q.; Mao, W.; et al. Pan-Cancer Analysis of NFE2L2 Mutations Identifies a Subset of Lung Cancers with Distinct Genomic and Improved Immunotherapy Outcomes. Cancer Cell Int. 2023, 23, 229. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhao, W.Q.; Fang, C.; Yang, X.; Ji, M. Histone Methyltransferase SETD2: A Potential Tumor Suppressor in Solid Cancers. J. Cancer 2020, 11, 3349–3356. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.K.; Hill, T.; Alexander, C.M. The Involvement of NRF2 in Lung Cancer. Oxidative Med. Cell. Longev. 2013, 2013, 746432. [Google Scholar] [CrossRef] [PubMed]
- Chanvorachote, P.; Sriratanasak, N.; Nonpanya, N. C-Myc Contributes to Malignancy of Lung Cancer: A Potential Anticancer Drug Target. Anticancer Res. 2020, 40, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, Y. Epithelial-to-Mesenchymal Transition and Its Role in EGFR-Mutant Lung Adenocarcinoma and Idiopathic Pulmonary Fibrosis. Pathol. Int. 2017, 67, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells by Transforming Growth Factor-1 Potential. Role in Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Camelo, A.; Dunmore, R.; Sleeman, M.A.; Clarke, D.L. The Epithelium in Idiopathic Pulmonary Fibrosis: Breaking the Barrier. Front. Pharmacol. 2014, 4, 173. [Google Scholar] [CrossRef]
- Schramm, F.; Schaefer, L.; Wygrecka, M. EGFR Signaling in Lung Fibrosis. Cells 2022, 11, 986. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S. Review of Epidermal Growth Factor Receptor Biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, S21–S26. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Sandhu, M.; Banerji, D.; Steri, V.; Olshen, A.; Moasser, M.M. The Role of HER2 and HER3 in HER2-Amplified Cancers beyond Breast Cancers. Sci. Rep. 2021, 11, 9091. [Google Scholar] [CrossRef] [PubMed]
- King, C.R.; Borrello, I.; Bellot, F.; Comoglio, P.; Schlessinger, J. Egf Binding to Its Receptor Triggers a Rapid Tyrosine Phosphorylation of the ErbB-2 Protein in the Mammary Tumor Cell Line SK-BR-3. EMBO J. 1988, 7, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Yu, D. Molecular Mechanisms of ErbB2-Mediated Breast Cancer Chemoresistance. Adv. Exp. Med. Biol. 2007, 608, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Korfhagen, T.R.; Swantz, R.J.; Wert, S.E.; Mccarty, J.M.; Kerlakian, C.B.; Glasser, S.W.; Whitsett, J.A. Respiratory Epithelial Cell Expression of Human Transforming Growth Factor-a Induces Lung Fibrosis in Transgenic Mice. J. Clin. Investig. 1994, 93, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Vallath, S.; Hynds, R.E.; Succony, L.; Janes, S.M.; Giangreco, A. Targeting EGFR Signalling in Chronic Lung Disease: Therapeutic Challenges and Opportunities. Eur. Respir. J. 2014, 44, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Andrianifahanana, M.; Wilkes, M.C.; Gupta, S.K.; Rahimi, R.A.; Repellin, C.E.; Edens, M.; Wittenberger, J.; Yin, X.; Maidl, E.; Becker, J.; et al. Profibrotic TGFβ Responses Require the Cooperative Action of PDGF and ErbB Receptor Tyrosine Kinases. FASEB J. 2013, 27, 4444–4454. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T.; Okudela, K.; Matsumura, M.; Baba, T.; Kitamura, H.; Arai, H.; Suzuki, T.; Koike, C.; Mutsui, H.; Sekiya, M.; et al. Significant Accumulation of KRAS Mutations in Bronchiolar Metaplasia-Associated Honeycomb Lesions of Interstitial Pneumonia. Oncol. Lett. 2022, 24, 225. [Google Scholar] [CrossRef]
- Guyard, A.; Danel, C.; Théou-Anton, N.; Debray, M.P.; Gibault, L.; Mordant, P.; Castier, Y.; Crestani, B.; Zalcman, G.; Blons, H.; et al. Morphologic and Molecular Study of Lung Cancers Associated with Idiopathic Pulmonary Fibrosis and Other Pulmonary Fibroses. Respir. Res. 2017, 18, 120. [Google Scholar] [CrossRef]
- Fujimoto, D.; Tomii, K.; Otoshi, T.; Kawamura, T.; Tamai, K.; Takeshita, J.; Tanaka, K.; Matsumoto, T.; Monden, K.; Nagata, K.; et al. Preexisting Interstitial Lung Disease Is Inversely Correlated to Tumor Epidermal Growth Factor Receptor Mutation in Patients with Lung Adenocarcinoma. Lung Cancer 2013, 80, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Sakashita, H.; Masai, K.; Totsuka, H.; Motoi, N.; Kobayashi, M.; Akashi, T.; Mimaki, S.; Tsuchihara, K.; Chiku, S.; et al. Deleterious Pulmonary Surfactant System Gene Mutations in Lung Adenocarcinomas Associated with Usual Interstitial Pneumonia. JCO Precis. Oncol. 2018, 2, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Shuman, A.G.; Aapro, M.S.; Anderson, B.; Arbour, K.; Barata, P.C.; Bardia, A.; Bruera, E.; Chabner, B.A.; Chen, H.; Choy, E.; et al. Supporting Patients with Cancer after Dobbs v. Jackson Women’s Health Organization. Oncologist 2022, 27, 711–713. [Google Scholar] [CrossRef]
- Ikeda, S.; Kato, T.; Kenmotsu, H.; Ogura, T.; Sato, Y.; Hino, A.; Harada, T.; Kubota, K.; Tokito, T.; Okamoto, I.; et al. Atezolizumab for Pretreated Non-Small Cell Lung Cancer with Idiopathic Interstitial Pneumonia: Final Analysis of Phase II AMBITIOUS Study. Oncologist 2022, 27, 720-e702. [Google Scholar] [CrossRef] [PubMed]
- Primiani, A.; Dias-Santagata, D.; John Iafrate, A.; Kradin, R.L. Pulmonary Adenocarcinoma Mutation Profile in Smokers with Smoking-Related Interstitial Fibrosis. Int. J. COPD 2014, 9, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Ikeda, S.; Tabata, E.; Kaneko, T.; Sagawa, S.; Yamada, C.; Kumagai, K.; Fukushima, T.; Haga, S.; Watanabe, M.; et al. KRASG12C Inhibitor as a Treatment Option for Non-Small-Cell Lung Cancer with Comorbid Interstitial Pneumonia. Cancers 2024, 16, 1327. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.A.; Kim, D.; Bae, S.H.; Chun, S.-M.; Song, J.S.; Kim, M.Y.; Song, J.W.; Kim, W.S.; Lee, J.C.; Park, S.; et al. MA05.05 Genomic Profiles of Lung Cancer Associated with Idiopathic Pulmonary Fibrosis. J. Thorac. Oncol. 2017, 12, S366. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Shimizu, J.; Oya, Y.; Watanabe, N.; Hasegawa, T.; Horio, Y.; Inaba, Y.; Fujiwara, Y. Risk Factors for Pneumonitis in Patients with Non-Small Cell Lung Cancer Treated with Immune Checkpoint Inhibitors plus Chemotherapy: A Retrospective Analysis. Thorac. Cancer 2022, 13, 724–731. [Google Scholar] [CrossRef]
- Johkoh, T.; Sakai, F.; Kusumoto, M.; Arakawa, H.; Harada, R.; Ueda, M.; Kudoh, S.; Fukuoka, M. Association between Baseline Pulmonary Status and Interstitial Lung Disease in Patients with Non-Small-Cell Lung Cancer Treated with Erlotinib—A Cohort Study. Clin. Lung Cancer 2014, 15, 448–454. [Google Scholar] [CrossRef]
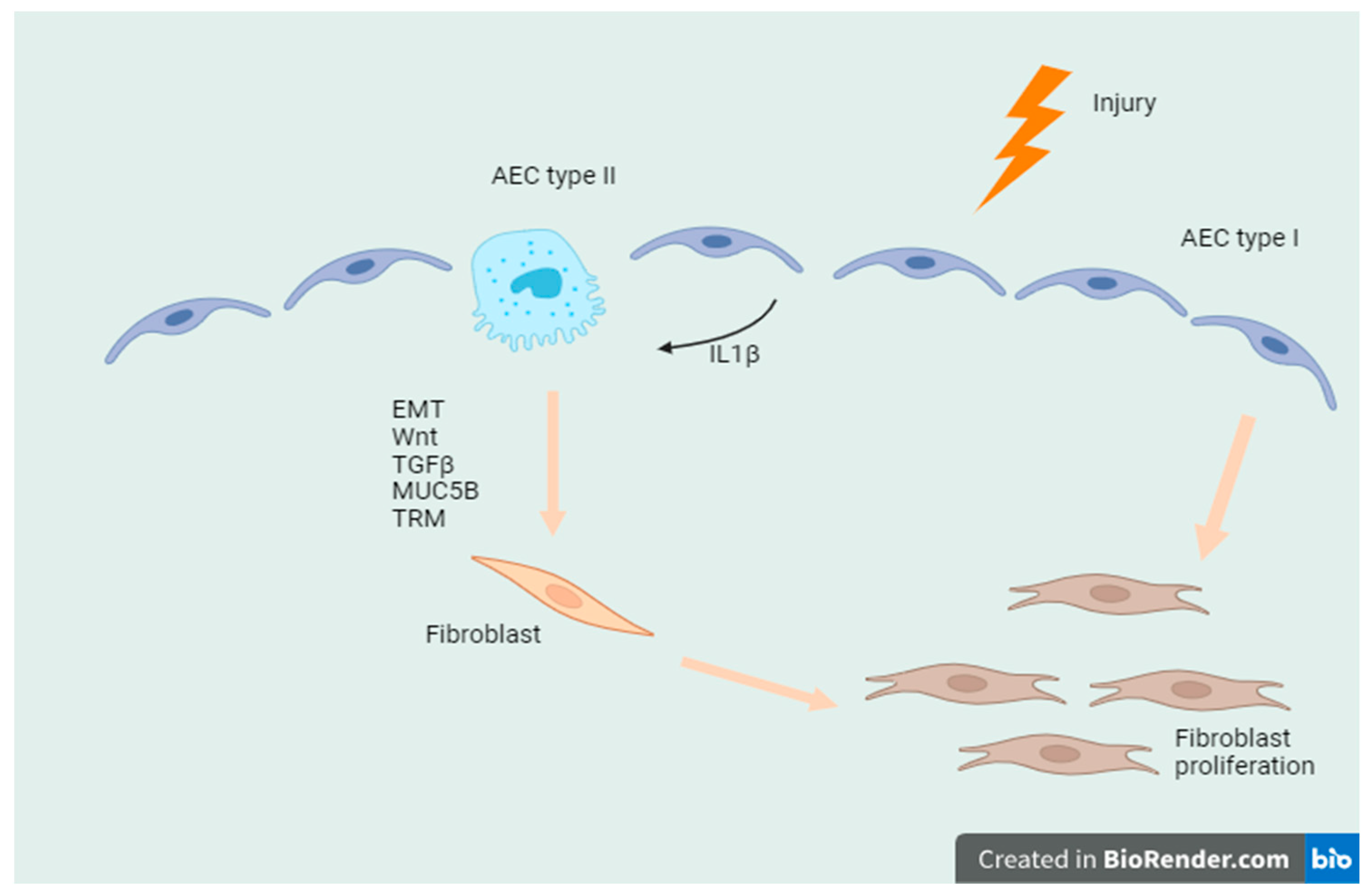
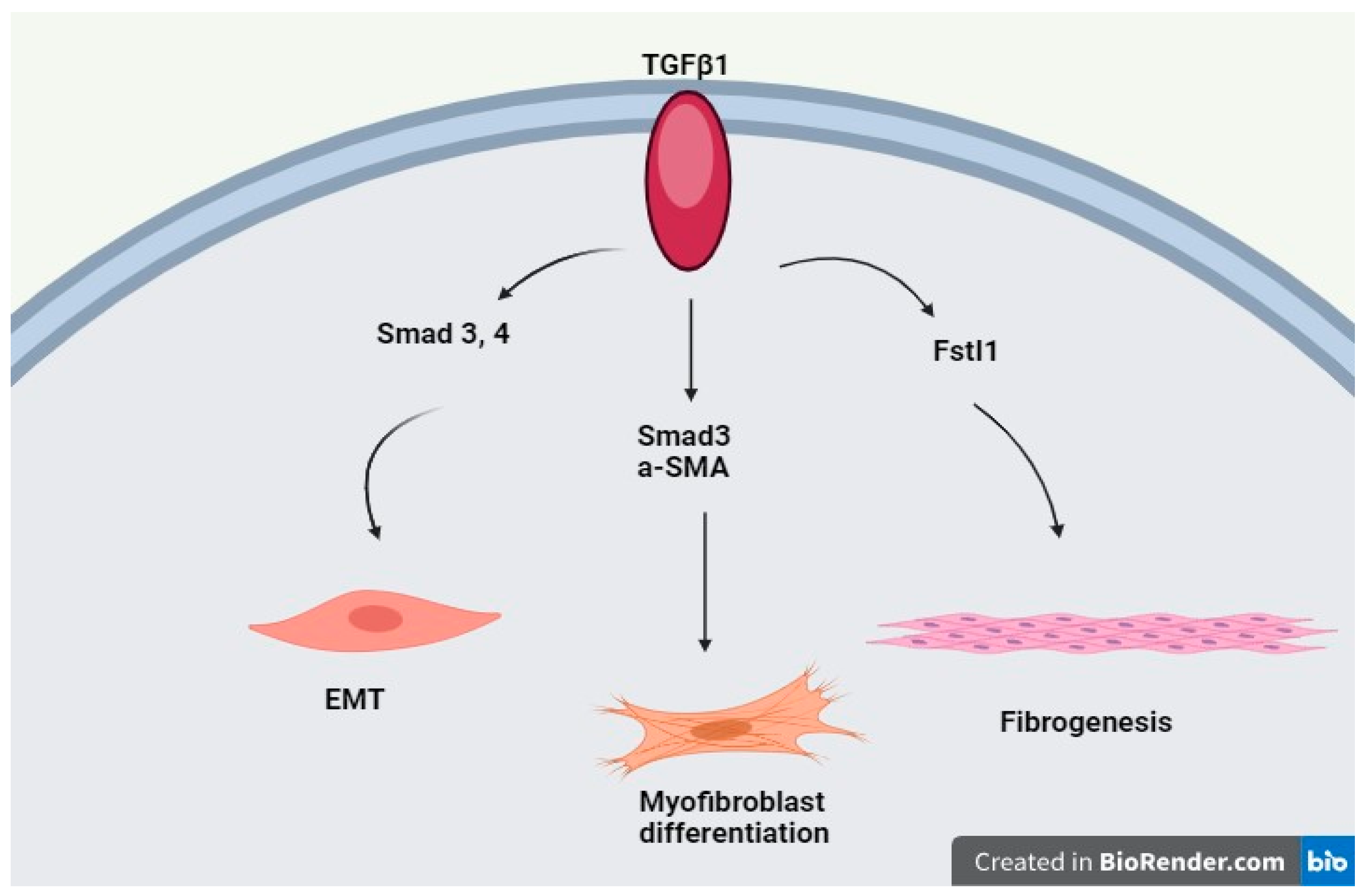

{kind=link}
{kind=link}
| Major | Idiopathic Pulmonary Fibrosis (IPF) |
| Idiopathic non-specific Interstitial pneumonia (NSIP) | |
| Respiratory bronchiolitis-interstitial lung disease (BR-ILD) | |
| Desquamative Interstitial pneumonia (DIP) | |
| Cryptogenic organization pneumonia (COP) | |
| Acute interstitial pneumonia (AIP) | |
| Rare | Idiopathic lymphoid interstitial pneumonia (LIP) |
| Idiopathic pleuro-parenchymatous fibroelastosis | |
| Unclassifiable | |
| New concepts | Interstitial pneumonia with autoimmune features (IPAF) |
| Progressive fibrosing interstitial lung diseases (PF-ILD) | |
| Interstitial lung abnormalities (ILAs) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sampsonas, F.; Bosgana, P.; Bravou, V.; Tzouvelekis, A.; Dimitrakopoulos, F.-I.; Kokkotou, E. Interstitial Lung Diseases and Non-Small Cell Lung Cancer: Particularities in Pathogenesis and Expression of Driver Mutations. Genes 2024, 15, 934. https://doi.org/10.3390/genes15070934
Sampsonas F, Bosgana P, Bravou V, Tzouvelekis A, Dimitrakopoulos F-I, Kokkotou E. Interstitial Lung Diseases and Non-Small Cell Lung Cancer: Particularities in Pathogenesis and Expression of Driver Mutations. Genes. 2024; 15(7):934. https://doi.org/10.3390/genes15070934
Chicago/Turabian StyleSampsonas, Fotios, Pinelopi Bosgana, Vasiliki Bravou, Argyrios Tzouvelekis, Foteinos-Ioannis Dimitrakopoulos, and Eleni Kokkotou. 2024. "Interstitial Lung Diseases and Non-Small Cell Lung Cancer: Particularities in Pathogenesis and Expression of Driver Mutations" Genes 15, no. 7: 934. https://doi.org/10.3390/genes15070934