
Diagnosis of Two Unrelated Syndromes of Prader-Willi and Calpainopathy: Insight from Trio Whole Genome Analysis and Isodisomy Mapping
 , , ,
, , , 

Abstract
1. Introduction
2. Participants, Materials, and Methods
2.1. Participants
2.2. Karyotyping
2.3. Methylation Specific-Multiplex Ligation-Dependent Probe Amplification
2.4. Whole Genome Sequencing
2.5. Zygosity and UPD Landscape Analysis by Trio WGS
2.6. UPD and Region of Homozygosity (ROH) Mapping
2.7. Focused Trio WGS Analysis on Muscular Dystrophy
2.8. Genome Variant Classification
3. Results
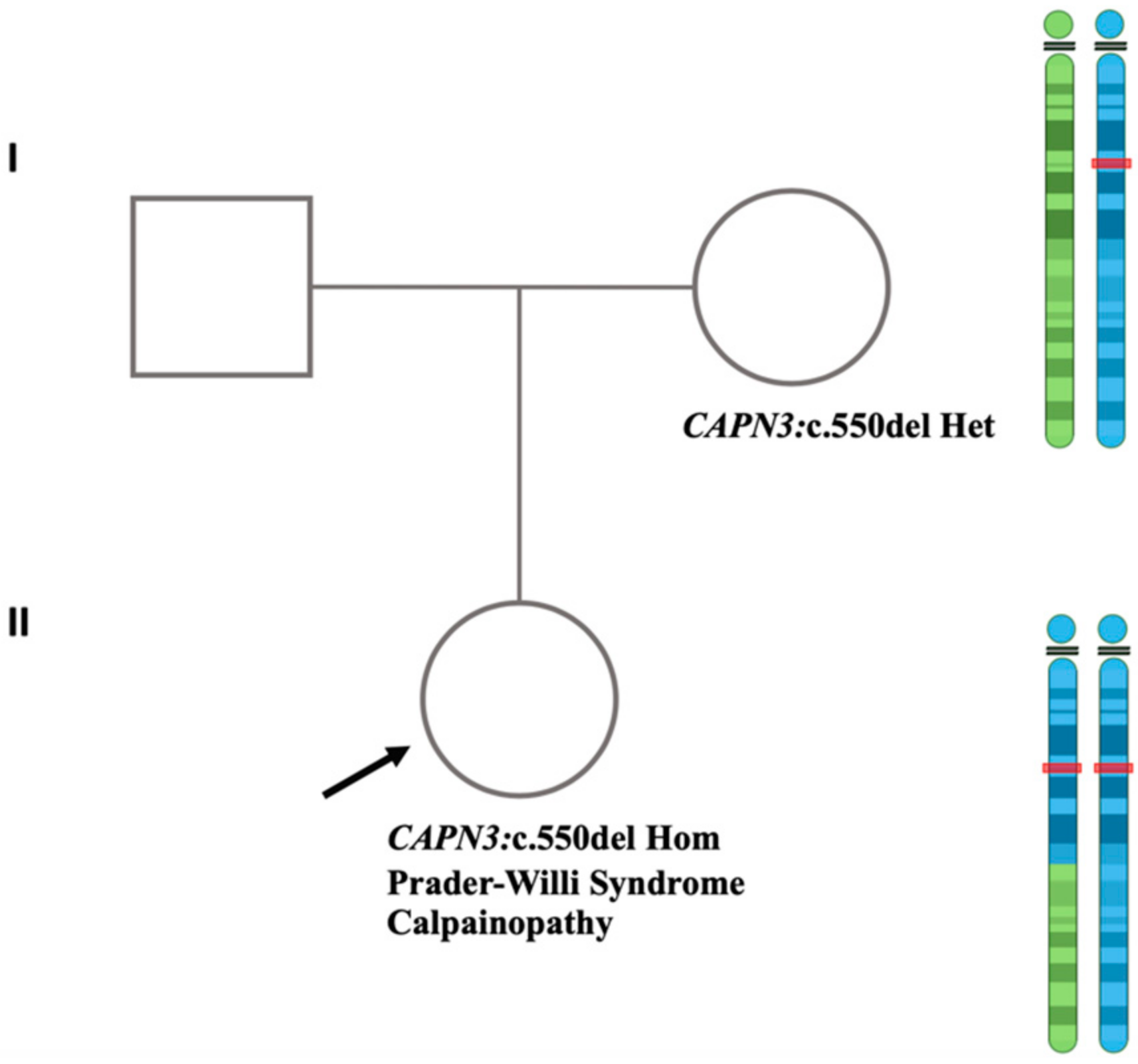
3.1. Clinical Findings
3.2. Genomic Findings
Karyotyping and Methylation Analysis
3.3. Ascertainment of UPD by Trio-WGS
3.4. Muscular Dystrophy Assessment Using Trio WGS Analysis
3.5. Assessment of the Breakpoint within UPD Region
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.E.; Rosenfeld, J.A.; James, R.A.; Bainbridge, M.; Niu, Z.; Wang, X.; Dhar, S.; Wiszniewski, W.; Akdemir, Z.H.; Gambin, T.; et al. Molecular diagnostic experience of whole-exome sequencing in adult patients. Genet. Med. 2016, 18, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Knapp, A.; Jagła, M.; Madetko-Talowska, A.; Szewczyk, K.; Książek, T.; Końska, K.; Kwinta, P. Paternal uniparental disomy of chromosome 2 resulting in a concurrent presentation of Crigler-Najjar syndrome type I and long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Am. J. Med. Genet. A 2022, 188, 1848–1852. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, Y.; Higashimoto, K.; Sasaki, K.; Jozaki, K.; Yoshinaga, H.; Okamoto, N.; Takama, Y.; Kubota, A.; Nakayama, M.; Yatsuki, H.; et al. Autosomal recessive cystinuria caused by genome-wide paternal uniparental isodisomy in a patient with Beckwith-Wiedemann syndrome. Clin. Genet. 2015, 88, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Benko, W.S.; Hruska, K.S.; Nagan, N.; Goker-Alpan, O.; Hart, P.S.; Schiffmann, R.; Sidransky, E. Uniparental disomy of chromosome 1 causing concurrent Charcot-Marie-Tooth and Gaucher disease Type 3. Neurology 2008, 70, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.B.; Forsythe, M.; Heeger, S.; Nicholls, R.D.; Schork, N.; Benn, P.; Schwartz, S. Comparison of phenotype between patients with Prader-Willi syndrome due to deletion 15q and uniparental disomy 15. Am. J. Med. Genet. 1997, 68, 433–440. [Google Scholar] [CrossRef]
- Gillessen-Kaesbach, G.; Robinson, W.; Lohmann, D.; Kaya-Westerloh, S.; Passarge, E.; Horsthemke, B. Genotype-phenotype correlation in a series of 167 deletion and non-deletion patients with Prader-Willi syndrome. Hum. Genet. 1995, 96, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, R.D.; Knoll, J.H.; Butler, M.G.; Karam, S.; Lalande, M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature 1989, 342, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Sorimachi, H.; Hata, S.; Ono, Y. Expanding members and roles of the calpain superfamily and their genetically modified animals. Exp. Anim. 2010, 59, 549–566. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Pajusalu, S.; Lake, N.J.; Zhou, G.; Ioannidis, N.; Mittal, P.; Johnson, N.E.; Weihl, C.C.; Williams, B.A.; Albrecht, D.E.; et al. Estimating prevalence for limb-girdle muscular dystrophy based on public sequencing databases. Genet. Med. 2019, 21, 2512–2520. [Google Scholar] [CrossRef]
- Nascimbeni, A.C.; Fanin, M.; Tasca, E.; Angelini, C. Transcriptional and translational effects of intronic CAPN3 gene mutations. Hum. Mutat. 2010, 31, E1658–E1669. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.L.; Angelini, C.; Daentl, D.; Garcia, C.; Greco, C.; Hausmanowa-Petrusewicz, I.; Fidzianska, A.; Wessel, H.; Hoffman, E.P. Calpain III mutation analysis of a heterogeneous limb-girdle muscular dystrophy population. Neurology 1999, 52, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Zatz, M.; Vainzof, M.; Passos-Bueno, M.R. Limb-girdle muscular dystrophy: One gene with different phenotypes, one phenotype with different genes. Curr. Opin. Neurol. 2000, 13, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Kramerova, I.; Beckmann, J.S.; Spencer, M.J. Molecular and cellular basis of calpainopathy (limb girdle muscular dystrophy type 2A). Biochim. Biophys. Acta 2007, 1772, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Richard, I.; Broux, O.; Allamand, V.; Fougerousse, F.; Chiannilkulchai, N.; Bourg, N.; Brenguier, L.; Devaud, C.; Pasturaud, P.; Roudaut, C. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995, 81, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Richard, I.; Brenguier, L.; Dinçer, P.; Roudaut, C.; Bady, B.; Burgunder, J.M.; Chemaly, R.; Garcia, C.A.; Halaby, G.; Jackson, C.E.; et al. Multiple independent molecular etiology for limb-girdle muscular dystrophy type 2A patients from various geographical origins. Am. J. Hum. Genet. 1997, 60, 1128–1138. [Google Scholar] [PubMed]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Cook, D.; Lawrence, M. ggbio: An R package for extending the grammar of graphics for genomic data. Genome Biol. 2012, 13, R77. [Google Scholar] [CrossRef]
- Nakka, P.; Pattillo Smith, S.; O’Donnell-Luria, A.H.; McManus, K.F.; Mountain, J.L.; Ramachandran, S.; Sathirapongsasuti, J.F.; 23andMe Research Team. Characterization of Prevalence and Health Consequences of Uniparental Disomy in Four Million Individuals from the General Population. Am. J. Hum. Genet. 2019, 105, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Polonis, K.; Lopes, J.L.; Cabral, H.; Babcock, H.E.; Kline, L.; Ruiz, K.M.; Schwartz, S.; Hasadsri, L.; Rowsey, R.A.; Hoppman, N.L. Uniparental disomy of multiple chromosomes in two cases with a complex phenotype. Am. J. Med. Genet. A 2023, 191, 1978–1983. [Google Scholar] [CrossRef] [PubMed]
- Molloy, B.; Jones, E.R.; Linhares, N.D.; Buckley, P.G.; Leahy, T.R.; Lynch, B.; Knerr, I.; King, M.D.; Gorman, K.M. Uniparental disomy screen of Irish rare disorder cohort unmasks homozygous variants of clinical significance in the TMCO1 and PRKRA genes. Front. Genet. 2022, 13, 945296. [Google Scholar] [CrossRef] [PubMed]
- Chien, S.C.; Chen, C.P.; Liou, J.D. Prenatal diagnosis and genetic counseling of uniparental disomy. Taiwan. J. Obstet. Gynecol. 2022, 61, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Kunta, A.R.; Jueng, J.; Jordan, C.; Kojic, J.; Mo, A.; Ebrahimi-Fakhari, D. Blended Phenotype of Prader-Willi Syndrome and HSP-SPG11 Caused by Maternal Uniparental Isodisomy. Neurol. Genet. 2022, 8, e200041. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, K.; Macke, E.L.; Klee, E.W.; Tebben, P.J.; Hand, J.L.; Hasadsri, L.; Marcou, C.A.; Schimmenti, L.A. Congenital ichthyosis in Prader-Willi syndrome associated with maternal chromosome 15 uniparental disomy: Case report and review of autosomal recessive conditions unmasked by UPD. Am. J. Med. Genet. A 2020, 182, 2442–2449. [Google Scholar] [CrossRef] [PubMed]
- Woodage, T.; Prasad, M.; Dixon, J.W.; Selby, R.E.; Romain, D.R.; Columbano-Green, L.M.; Graham, D.; Rogan, P.K.; Seip, J.R.; Smith, A. Bloom syndrome and maternal uniparental disomy for chromosome 15. Am. J. Hum. Genet. 1994, 55, 74–80. [Google Scholar] [PubMed]
- Zeesman, S.; McCready, E.; Sadikovic, B.; Nowaczyk, M.J. Prader-Willi syndrome and Tay-Sachs disease in association with mixed maternal uniparental isodisomy and heterodisomy 15 in a girl who also had isochromosome Xq. Am. J. Med. Genet. A 2015, 167A, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Lynn, C.H.; Driscoll, D.C.; Goldstone, A.P.; Gold, J.A.; Kimonis, V.; Dykens, E.; Butler, M.G.; Shuster, J.J.; Driscoll, D.J. Nutritional phases in Prader-Willi syndrome. Am. J. Med. Genet. A 2011, 155A, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Sturich, J.; Myers, S.E.; Gold, J.A.; Kimonis, V.; Driscoll, D.J. Is gestation in Prader-Willi syndrome affected by the genetic subtype? J. Assist. Reprod. Genet. 2009, 26, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Investig. 2015, 38, 1249–1263. [Google Scholar] [CrossRef]
- Ohno, S.; Minoshima, S.; Kudoh, J.; Fukuyama, R.; Shimizu, Y.; Ohmi-Imajoh, S.; Shimizu, N.; Suzuki, K. Four genes for the calpain family locate on four distinct human chromosomes. Cytogenet. Cell Genet. 1990, 53, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Sorimachi, H.; Imajoh-Ohmi, S.; Emori, Y.; Kawasaki, H.; Ohno, S.; Minami, Y.; Suzuki, K. Molecular cloning of a novel mammalian calcium-dependent protease distinct from both m- and mu-types. Specific expression of the mRNA in skeletal muscle. J. Biol. Chem. 1989, 264, 20106–20111. [Google Scholar] [CrossRef] [PubMed]
- Hata, S.; Doi, N.; Shinkai-Ouchi, F.; Ono, Y. A muscle-specific calpain, CAPN3, forms a homotrimer. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140411. [Google Scholar] [CrossRef] [PubMed]
- Baghdiguian, S.; Martin, M.; Richard, I.; Pons, F.; Astier, C.; Bourg, N.; Hay, R.T.; Chemaly, R.; Halaby, G.; Loiselet, J.; et al. Calpain 3 deficiency is associated with myonuclear apoptosis and profound perturbation of the IkappaB α/NF-kappaB pathway in limb-girdle muscular dystrophy type 2A. Nat. Med. 1999, 5, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Sorimachi, H.; Kinbara, K.; Kimura, S.; Takahashi, M.; Ishiura, S.; Sasagawa, N.; Sorimachi, N.; Shimada, H.; Tagawa, K.; Maruyama, K. Muscle-specific calpain, p94, responsible for limb girdle muscular dystrophy type 2A, associates with connectin through IS2, a p94-specific sequence. J. Biol. Chem. 1995, 270, 31158–31162. [Google Scholar] [CrossRef]
- Taveau, M.; Bourg, N.; Sillon, G.; Roudaut, C.; Bartoli, M.; Richard, I. Calpain 3 is activated through autolysis within the active site and lyses sarcomeric and sarcolemmal components. Mol. Cell. Biol. 2003, 23, 9127–9135. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Tang, F.; Gao, H.; Zhang, X.; Li, X.; Xiao, D. CAPN3: A muscle-specific calpain with an important role in the pathogenesis of diseases (Review). Int. J. Mol. Med. 2021, 48, 203. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Zheng, Y.; Zhao, Z.; Lin, P.; Xi, J.; Zhu, W.; Lin, J.; Lu, J.; Yu, M.; Zhang, W.; et al. Molecular landscape of CAPN3 mutations in limb-girdle muscular dystrophy type R1: From a Chinese multicentre analysis to a worldwide perspective. J. Med. Genet. 2021, 58, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Canki-Klain, N.; Milic, A.; Kovac, B.; Trlaja, A.; Grgicevic, D.; Zurak, N.; Fardeau, M.; Leturcq, F.; Kaplan, J.C.; Urtizberea, J.A.; et al. Prevalence of the 550delA mutation in calpainopathy (LGMD 2A) in Croatia. Am. J. Med. Genet. A 2004, 125A, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Topaloğlu, H.; Dinçer, P.; Richard, I.; Akçören, Z.; Alehan, D.; Ozme, S.; Cağlar, M.; Karaduman, A.; Urtizberea, J.A.; Beckmann, J.S. Calpain-3 deficiency causes a mild muscular dystrophy in childhood. Neuropediatrics 1997, 28, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Dinçer, P.; Leturcq, F.; Richard, I.; Piccolo, F.; Yalnizoglu, D.; de Toma, C.; Akçören, Z.; Broux, O.; Deburgrave, N.; Brenguier, L.; et al. A biochemical, genetic, and clinical survey of autosomal recessive limb girdle muscular dystrophies in Turkey. Ann. Neurol. 1997, 42, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, T.V.; Krakhmaleva, I.N.; Lipatova, N.A.; Shakhovskaya, N.I.; Shishkin, S.S.; Limborska, S.A. High incidence of 550delA mutation of CAPN3 in LGMD2 patients from Russia. Hum. Mutat. 2000, 15, 295. [Google Scholar] [CrossRef]
- Yamazawa, K.; Ogata, T.; Ferguson-Smith, A.C. Uniparental disomy and human disease: An overview. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Del Gaudio, D.; Shinawi, M.; Astbury, C.; Tayeh, M.K.; Deak, K.L.; Raca, G.; Committee, A.L.Q.A. Diagnostic testing for uniparental disomy: A points to consider statement from the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2020, 22, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Benn, P. Uniparental disomy: Origin, frequency, and clinical significance. Prenat. Diagn. 2021, 41, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Koehler, K.E.; Hawley, R.S.; Sherman, S.; Hassold, T. Recombination and nondisjunction in humans and flies. Hum. Mol. Genet. 1996, 5, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Guissart, C.; Latypova, X.; Rollier, P.; Khan, T.N.; Stamberger, H.; McWalter, K.; Cho, M.T.; Kjaergaard, S.; Weckhuysen, S.; Lesca, G.; et al. Dual Molecular Effects of Dominant RORA Mutations Cause Two Variants of Syndromic Intellectual Disability with Either Autism or Cerebellar Ataxia. Am. J. Hum. Genet. 2018, 102, 744–759. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytoband | Size | Location | UPD Type |
|---|---|---|---|
| 15q11.2–15q22.2 | 38.61 Mbp | Chr15: 22373341-60984740 | Mat Isodisomy |
| 15q22.2–15q26.3 | 41.55 Mbp | Chr15: 60984741-102531392 | Mat Heterodisomy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuk, M.; Unal, B.; Bevanda, A.; Hayes, C.P.; Walker, M.; Abraamyan, F.; Beluzic, R.; Gornik, K.C.; Ozretic, D.; Prutki, M.; et al. Diagnosis of Two Unrelated Syndromes of Prader-Willi and Calpainopathy: Insight from Trio Whole Genome Analysis and Isodisomy Mapping. Genes 2024, 15, 946. https://doi.org/10.3390/genes15070946
Cuk M, Unal B, Bevanda A, Hayes CP, Walker M, Abraamyan F, Beluzic R, Gornik KC, Ozretic D, Prutki M, et al. Diagnosis of Two Unrelated Syndromes of Prader-Willi and Calpainopathy: Insight from Trio Whole Genome Analysis and Isodisomy Mapping. Genes. 2024; 15(7):946. https://doi.org/10.3390/genes15070946
Chicago/Turabian StyleCuk, Mario, Busra Unal, Andjela Bevanda, Connor P. Hayes, McKenzie Walker, Feruza Abraamyan, Robert Beluzic, Kristina Crkvenac Gornik, David Ozretic, Maja Prutki, and et al. 2024. "Diagnosis of Two Unrelated Syndromes of Prader-Willi and Calpainopathy: Insight from Trio Whole Genome Analysis and Isodisomy Mapping" Genes 15, no. 7: 946. https://doi.org/10.3390/genes15070946
APA StyleCuk, M., Unal, B., Bevanda, A., Hayes, C. P., Walker, M., Abraamyan, F., Beluzic, R., Gornik, K. C., Ozretic, D., Prutki, M., Nie, Q., Reddi, H. V., & Ghazani, A. A. (2024). Diagnosis of Two Unrelated Syndromes of Prader-Willi and Calpainopathy: Insight from Trio Whole Genome Analysis and Isodisomy Mapping. Genes, 15(7), 946. https://doi.org/10.3390/genes15070946