
The Missense Variant in the Signal Peptide of α-GLA Gene, c.13 A/G, Promotes Endoplasmic Reticular Stress and the Related Pathway’s Activation

 ,
,  ,
,  , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Genetic Analysis
2.2. Cell Culture
2.3. Plasmid Amplification and Purification
2.4. Transient Transfection
2.5. mRNA Extraction and Reverse Transcription–Polymerase Chain Reaction (RT-PCR)
2.6. Protein Extraction and Western Blotting Analysis
2.7. Transmission Electron Microscopy (TEM)
2.8. Peripheral Blood Mononuclear Cell (PBMC) Isolation
2.9. Statistical Analysis
3. Results
3.1. Clinical and Biochemical Features of the Proband
3.2. In Vitro Transfection of the Genetic Variant c.13 A/G, Codifying the Signal Peptide
3.3. The Genetic Variant c.13 A/G Promotes ER Injury and Intra-Lysosomal Accumulation
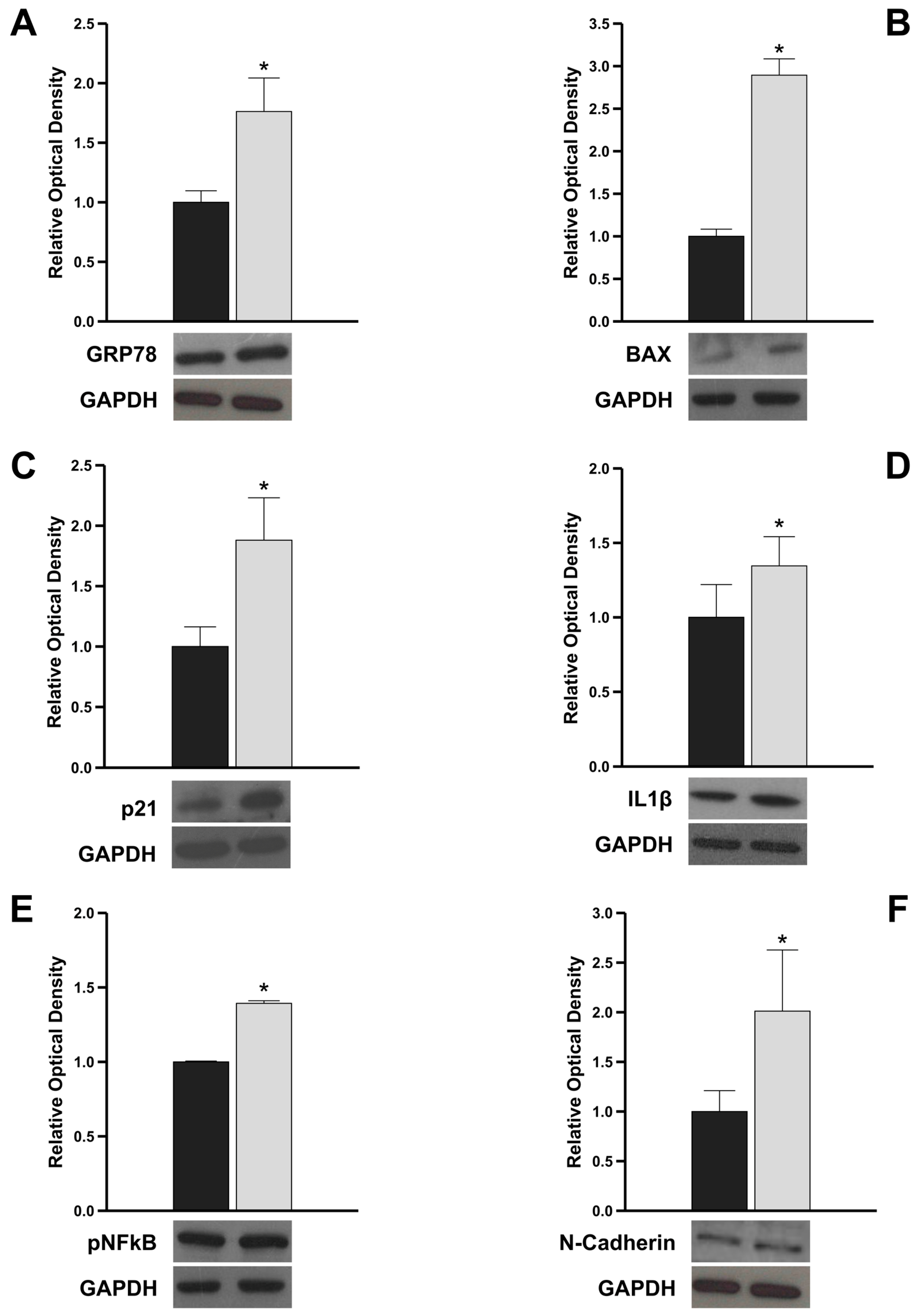
3.4. The Genetic Variant c.13 A/G Triggers Pro-Apoptotic, Pro-Inflammatory, and Pro-Fibrotic Intracellular Signals by Increasing GRP78 Expression
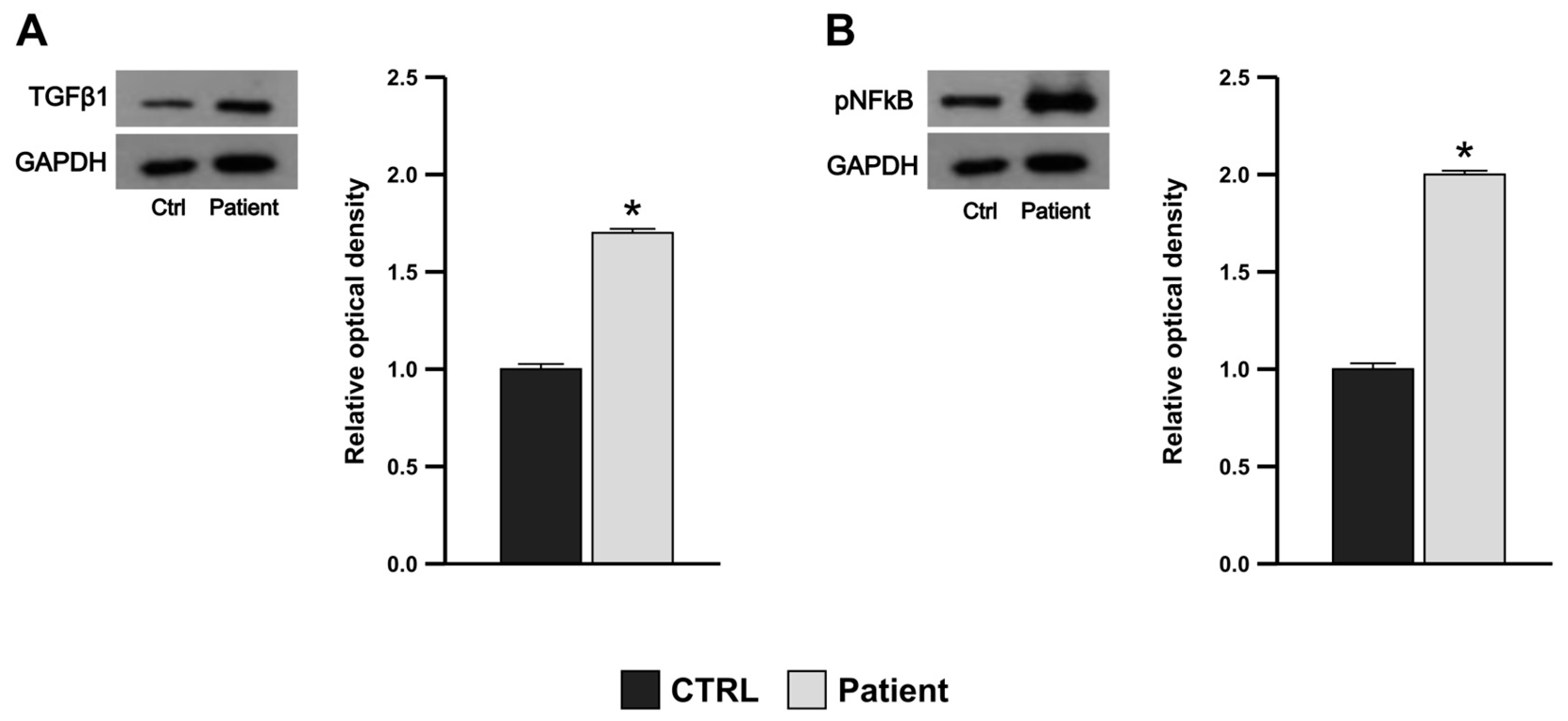
3.5. Proband’s PBMCs Exhibit Pro-Inflammatory and Pro-Fibrotic Profile
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Germain, D.P. Fabry Disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Simonetta, I.; Riolo, R.; Todaro, F.; Di Chiara, T.; Miceli, S.; Pinto, A. Pathogenesis and Molecular Mechanisms of Anderson-Fabry Disease and Possible New Molecular Addressed Therapeutic Strategies. Int. J. Mol. Sci. 2021, 22, 10088. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld, P.; Feriozzi, S. Contribution of Inflammatory Pathways to Fabry Disease Pathogenesis. Mol. Genet. Metab. 2017, 122, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.T.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD): 2003 Update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef] [PubMed]
- van der Tol, L.; Smid, B.E.; Poorthuis, B.J.H.M.; Biegstraaten, M.; Deprez, R.H.L.; Linthorst, G.E.; Hollak, C.E.M. A Systematic Review on Screening for Fabry Disease: Prevalence of Individuals with Genetic Variants of Unknown Significance. J. Med. Genet. 2014, 51, 1–9. [Google Scholar] [CrossRef]
- Paetzel, M.; Karla, A.; Strynadka, N.C.J.; Dalbey, R.E. Signal Peptidases. Chem. Rev. 2002, 102, 4549–4580. [Google Scholar] [CrossRef] [PubMed]
- Gemmer, M.; Chaillet, M.L.; van Loenhout, J.; Cuevas Arenas, R.; Vismpas, D.; Gröllers-Mulderij, M.; Koh, F.A.; Albanese, P.; Scheltema, R.A.; Howes, S.C.; et al. Visualization of Translation and Protein Biogenesis at the ER Membrane. Nature 2023, 614, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Li, X.; Mariappan, M. Signal Sequences Encode Information for Protein Folding in the Endoplasmic Reticulum. J. Cell Biol. 2023, 222, e202203070. [Google Scholar] [CrossRef]
- Beena, K.; Udgaonkar, J.B.; Varadarajan, R. Effect of Signal Peptide on the Stability and Folding Kinetics of Maltose Binding Protein. Biochemistry 2004, 43, 3608–3619. [Google Scholar] [CrossRef]
- Wu, J.-L.; Chen, Y.-R. Signal Peptide Stabilizes Folding and Inhibits Misfolding of Serum Amyloid A. Protein Sci. 2022, 31, e4485. [Google Scholar] [CrossRef]
- Smets, D.; Smit, J.; Xu, Y.; Karamanou, S.; Economou, A. Signal Peptide-Rheostat Dynamics Delay Secretory Preprotein Folding. J. Mol. Biol. 2022, 434, 167790. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the Unfolded Protein Response Regulator GRP78/BiP in Development, Cancer, and Neurological Disorders. Antioxid. Redox Signal 2009, 11, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A Cell’s Response to Stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of Endoplasmic Reticulum Stress-Induced Apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Kaczmarek, A.; Krysko, O.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. ER Stress-Induced Inflammation: Does It Aid or Impede Disease Progression? Trends Mol. Med. 2012, 18, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Morrone, A.; Cavicchi, C.; Bardelli, T.; Antuzzi, D.; Parini, R.; Di Rocco, M.; Feriozzi, S.; Gabrielli, O.; Barone, R.; Pistone, G.; et al. Fabry Disease: Molecular Studies in Italian Patients and X Inactivation Analysis in Manifesting Carriers. J. Med. Genet. 2003, 40, e103. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Xue, H.; Pritchard, H.W.; Wang, X. Reactive Oxygen Species-Provoked Mitochondria-Dependent Cell Death during Ageing of Elm (Ulmus pumila L.) Seeds. Plant J. 2015, 81, 438–452. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Topping, T.B.; Randall, L.L. Physiological Role during Export for the Retardation of Folding by the Leader Peptide of Maltose-Binding Protein. Proc. Natl. Acad. Sci. USA 1989, 86, 9213–9217. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. The Mammalian Unfolded Protein Response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.-K. An Update on the Regulatory Mechanisms of NLRP3 Inflammasome Activation. Cell Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef]
- Debnath, P.; Huirem, R.S.; Dutta, P.; Palchaudhuri, S. Epithelial-Mesenchymal Transition and Its Transcription Factors. Biosci. Rep. 2022, 42, BSR20211754. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, P.N.; Mucci, J.M.; Ceci, R.; Fossati, C.A.; Rozenfeld, P.A. Higher Apoptotic State in Fabry Disease Peripheral Blood Mononuclear Cells.: Effect of Globotriaosylceramide. Mol. Genet. Metab. 2011, 104, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Braunstein, H.; Papazian, M.; Maor, G.; Lukas, J.; Rolfs, A.; Horowitz, M. Misfolding of Lysosomal α-Galactosidase a in a Fly Model and Its Alleviation by the Pharmacological Chaperone Migalastat. Int. J. Mol. Sci. 2020, 21, 7397. [Google Scholar] [CrossRef] [PubMed]
- Consolato, F.; De Fusco, M.; Schaeffer, C.; Pieruzzi, F.; Scolari, F.; Gallieni, M.; Lanzani, C.; Feriozzi, S.; Rampoldi, L. α-Gal A Missense Variants Associated with Fabry Disease Can Lead to ER Stress and Induction of the Unfolded Protein Response. Mol. Genet. Metab. Rep. 2022, 33, 100926. [Google Scholar] [CrossRef] [PubMed]
- Oder, D.; Vergho, D.; Ertl, G.; Wanner, C.; Nordbeck, P. Case Report of a 45-Year Old Female Fabry Disease Patient Carrying Two Alpha-Galactosidase A Gene Mutation Alleles. BMC Med. Genet. 2016, 17, 46. [Google Scholar] [CrossRef]
- Lavalle, L.; Thomas, A.S.; Beaton, B.; Ebrahim, H.; Reed, M.; Ramaswami, U.; Elliott, P.; Mehta, A.B.; Hughes, D.A. Phenotype and Biochemical Heterogeneity in Late Onset Fabry Disease Defined by N215S Mutation. PLoS ONE 2018, 13, e0193550. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bossio, S.; Perrotta, I.D.; Lofaro, D.; La Russa, D.; Rago, V.; Bonofiglio, R.; Greco, R.; Andreucci, M.; Aversa, A.; La Russa, A.; et al. The Missense Variant in the Signal Peptide of α-GLA Gene, c.13 A/G, Promotes Endoplasmic Reticular Stress and the Related Pathway’s Activation. Genes 2024, 15, 947. https://doi.org/10.3390/genes15070947
Bossio S, Perrotta ID, Lofaro D, La Russa D, Rago V, Bonofiglio R, Greco R, Andreucci M, Aversa A, La Russa A, et al. The Missense Variant in the Signal Peptide of α-GLA Gene, c.13 A/G, Promotes Endoplasmic Reticular Stress and the Related Pathway’s Activation. Genes. 2024; 15(7):947. https://doi.org/10.3390/genes15070947
Chicago/Turabian StyleBossio, Sabrina, Ida Daniela Perrotta, Danilo Lofaro, Daniele La Russa, Vittoria Rago, Renzo Bonofiglio, Rosita Greco, Michele Andreucci, Antonio Aversa, Antonella La Russa, and et al. 2024. "The Missense Variant in the Signal Peptide of α-GLA Gene, c.13 A/G, Promotes Endoplasmic Reticular Stress and the Related Pathway’s Activation" Genes 15, no. 7: 947. https://doi.org/10.3390/genes15070947
APA StyleBossio, S., Perrotta, I. D., Lofaro, D., La Russa, D., Rago, V., Bonofiglio, R., Greco, R., Andreucci, M., Aversa, A., La Russa, A., & Perri, A. (2024). The Missense Variant in the Signal Peptide of α-GLA Gene, c.13 A/G, Promotes Endoplasmic Reticular Stress and the Related Pathway’s Activation. Genes, 15(7), 947. https://doi.org/10.3390/genes15070947