


Exploring the Genotype–Phenotype Correlations in a Child with Inherited Seizure and Thrombocytopenia by Digenic Network Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient
2.2. Whole Exome Sequencing (WES) and Sanger Validation
2.3. Variant Analysis and Interpretation
2.4. Digenic Network Analysis
2.5. Protein–Protein Interactions and Pathways Analysis
2.6. Gene Expression and Co-Expression Analysis
3. Results
3.1. Clinical Observations
3.2. WES and Variants Interpretation
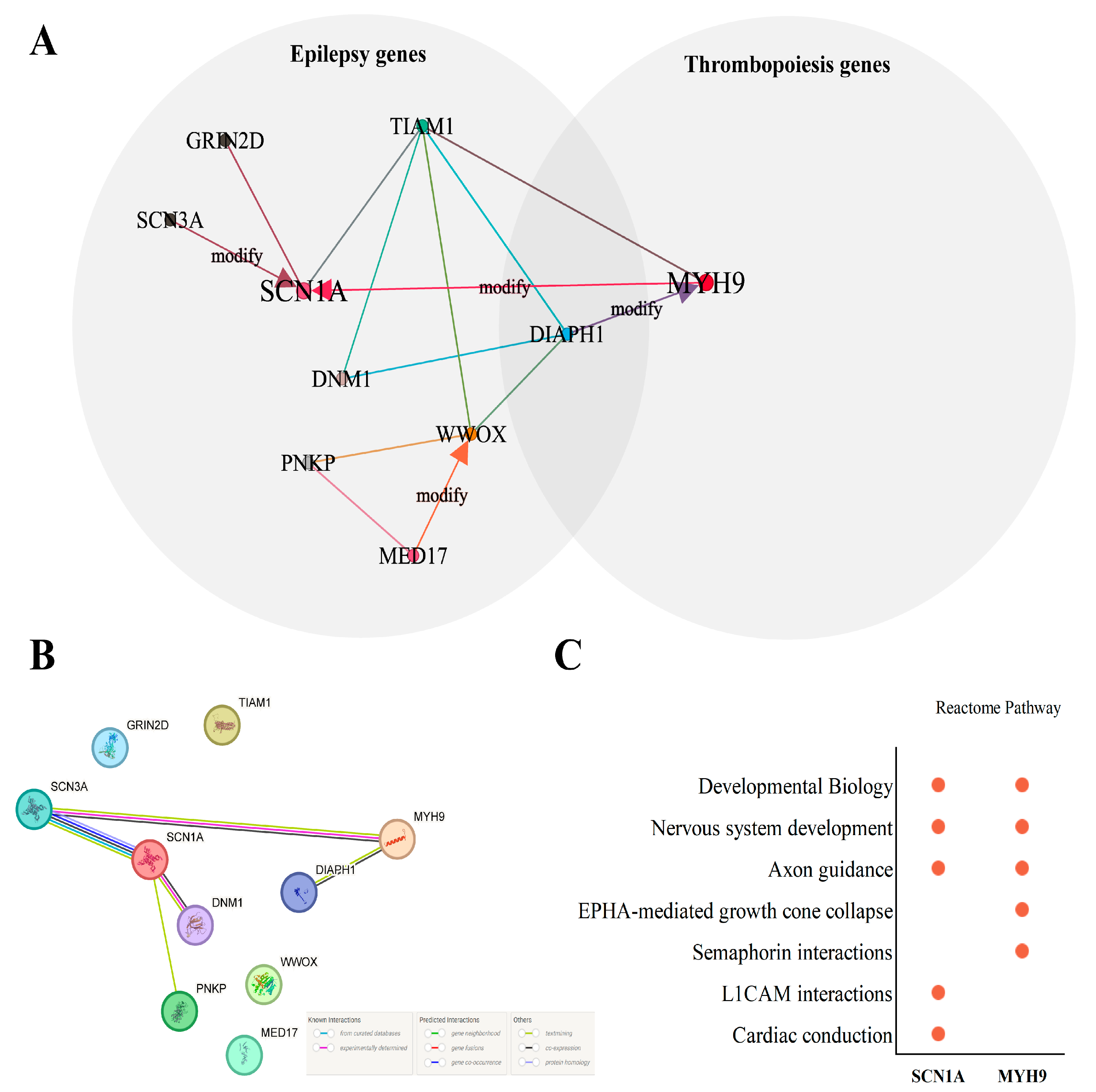
3.3. Digenic Network and Combinations Annotation
3.4. Protein–Protein Interactions and Pathways Analysis of Core Genes
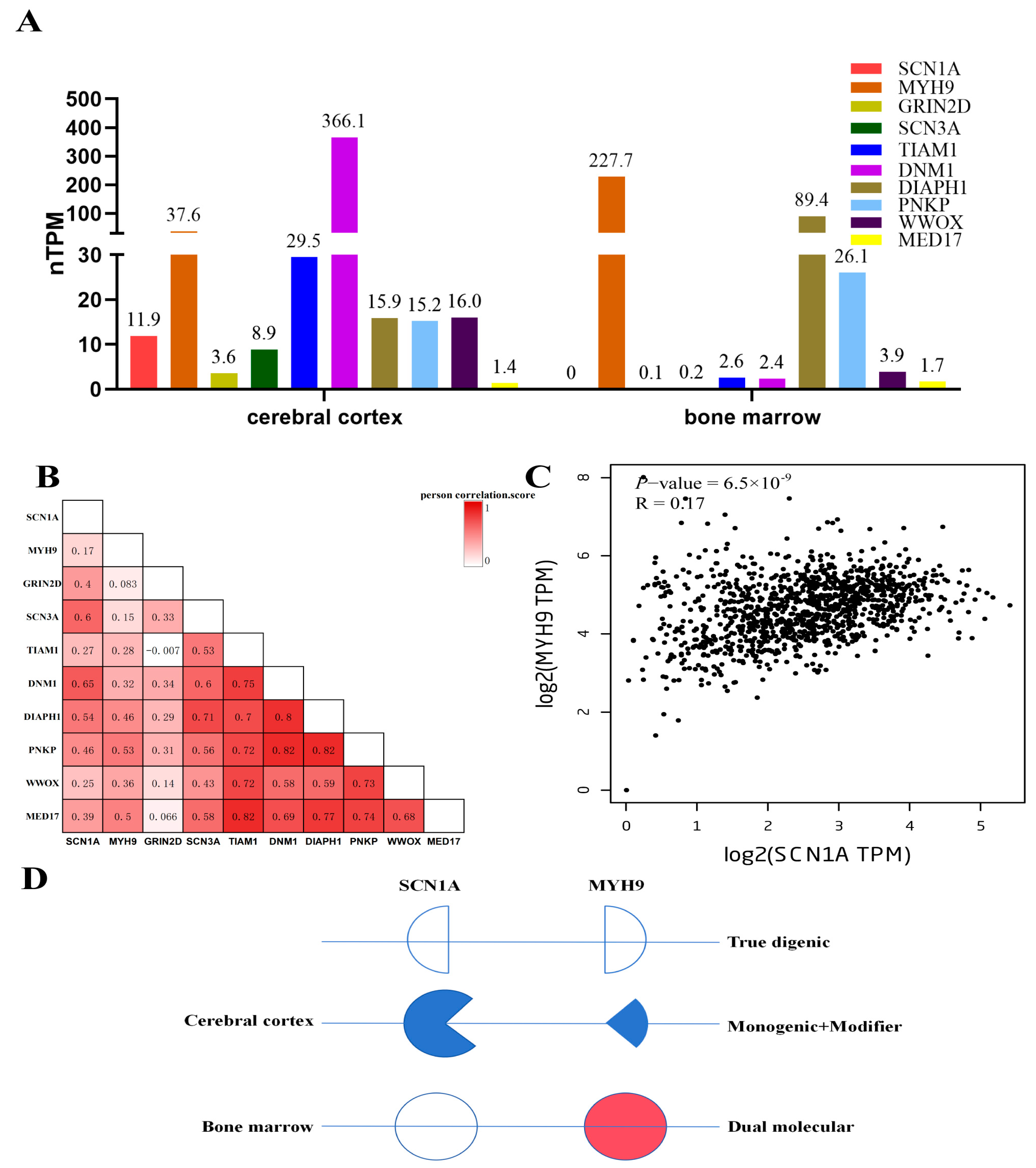
3.5. Expression, Co-Expression Profiles of Genes, and Digenic Model for Patient
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chong, J.X.; Buckingham, K.J.; Jhangiani, S.N.; Boehm, C.; Sobreira, N.; Smith, J.D.; Harrell, T.M.; McMillin, M.J.; Wiszniewski, W.; Gambin, T.; et al. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am. J. Hum. Genet. 2015, 97, 199–215. [Google Scholar] [CrossRef]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Akdemir, Z.H.C.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef]
- Gazzo, A.; Raimondi, D.; Daneels, D.; Moreau, Y.; Smits, G.; Van Dooren, S.; Lenaerts, T. Understanding mutational effects in digenic diseases. Nucleic Acids Res. 2017, 45, e140. [Google Scholar] [CrossRef]
- Papadimitriou, S.; Gazzo, A.; Versbraegen, N.; Nachtegael, C.; Aerts, J.; Moreau, Y.; Van Dooren, S.; Nowé, A.; Smits, G.; Lenaerts, T. Predicting disease-causing variant combinations. Proc. Natl. Acad. Sci. USA 2019, 116, 11878–11887. [Google Scholar] [CrossRef]
- Deltas, C. Digenic inheritance and genetic modifiers. Clin. Genet. 2018, 93, 429–438. [Google Scholar] [CrossRef]
- Schäffer, A.A. Digenic inheritance in medical genetics. J. Med. Genet. 2013, 50, 641–652. [Google Scholar] [CrossRef]
- Rahit, K.M.T.H.; Tarailo-Graovac, M. Genetic Modifiers and Rare Mendelian Disease. Genes 2020, 11, 239. [Google Scholar] [CrossRef]
- van Heyningen, V.; Yeyati, P.L. Mechanisms of non-Mendelian inheritance in genetic disease. Hum. Mol. Genet. 2004, 13, R225–R233. [Google Scholar] [CrossRef]
- Badano, J.L.; Katsanis, N. Beyond Mendel: An evolving view of human genetic disease transmission. Nat. Rev. Genet. 2002, 3, 779–789. [Google Scholar] [CrossRef]
- Riordan, J.D.; Nadeau, J.H. From Peas to Disease: Modifier Genes, Network Resilience, and the Genetics of Health. Am. J. Hum. Genet. 2017, 101, 177–191. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Nabbout, R. SCN1A-related phenotypes: Epilepsy and beyond. Epilepsia 2019, 60 (Suppl. S3), S17–S24. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, C.; Petrelli, C.; Mei, D.; Foschi, N.; Guerrini, R.; Provinciali, L.; Zamponi, N. A novel inherited SCN1A mutation associated with different neuropsychological phenotypes: Is there a common core deficit? Epilepsy Behav. 2015, 43, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, R.; Cellini, E.; Mei, D.; Metitieri, T.; Petrelli, C.; Pucatti, D.; Marini, C.; Zamponi, N. Variable epilepsy phenotypes associated with a familial intragenic deletion of the SCN1A gene. Epilepsia 2010, 51, 2474–2477. [Google Scholar] [CrossRef] [PubMed]
- Pecci, A.; Ma, X.; Savoia, A.; Adelstein, R.S. MYH9: Structure, functions and role of non-muscle myosin IIA in human disease. Gene 2018, 664, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Asensio-Juárez, G.; Llorente-González, C.; Vicente-Manzanares, M. Linking the Landscape of MYH9-Related Diseases to the Molecular Mechanisms that Control Non-Muscle Myosin II-A Function in Cells. Cells 2020, 9, 1458. [Google Scholar] [CrossRef] [PubMed]
- Pecci, A.; Klersy, C.; Gresele, P.; Lee, K.J.; De Rocco, D.; Bozzi, V.; Russo, G.; Heller, P.G.; Loffredo, G.; Ballmaier, M.; et al. MYH9-related disease: A novel prognostic model to predict the clinical evolution of the disease based on genotype-phenotype correlations. Hum. Mutat. 2014, 35, 236–247. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Stephenson, J.D.; Sillitoe, I.; Orengo, C.A.; Thornton, J.M. VarSite: Disease variants and protein structure. Protein Sci. 2020, 29, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Renaux, A.; Papadimitriou, S.; Versbraegen, N.; Nachtegael, C.; Boutry, S.; Nowé, A.; Smits, G.; Lenaerts, T. ORVAL: A novel platform for the prediction and exploration of disease-causing oligogenic variant combinations. Nucleic Acids Res. 2019, 47, W93–W98. [Google Scholar] [CrossRef]
- Chi, W.; Kiskinis, E. Integrative analysis of epilepsy-associated genes reveals expression-phenotype correlations. Sci. Rep. 2024, 14, 3587. [Google Scholar] [CrossRef]
- Kim, B. Diagnostic workup of inherited platelet disorders. Blood Res. 2022, 57, S11–S19. [Google Scholar] [CrossRef]
- Versbraegen, N.; Gravel, B.; Nachtegael, C.; Renaux, A.; Verkinderen, E.; Nowé, A.; Lenaerts, T.; Papadimitriou, S. Faster and more accurate pathogenic combination predictions with VarCoPP2.0. BMC Bioinform. 2023, 24, 179. [Google Scholar] [CrossRef]
- Versbraegen, N.; Fouché, A.; Nachtegael, C.; Papadimitriou, S.; Gazzo, A.; Smits, G.; Lenaerts, T. Using game theory and decision decomposition to effectively discern and characterise bi-locus diseases. Artif. Intell. Med. 2019, 99, 101690. [Google Scholar] [CrossRef] [PubMed]
- Gazzo, A.M.; Daneels, D.; Cilia, E.; Bonduelle, M.; Abramowicz, M.; Van Dooren, S.; Smits, G.; Lenaerts, T. DIDA: A curated and annotated digenic diseases database. Nucleic Acids Res. 2016, 44, D900–D907. [Google Scholar] [CrossRef]
- Nachtegael, C.; Gravel, B.; Dillen, A.; Smits, G.; Nowé, A.; Papadimitriou, S.; Lenaerts, T. Scaling up oligogenic diseases research with OLIDA: The Oligogenic Diseases Database. Database 2022, 2022, baac023. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Milacic, M.; Beavers, D.; Conley, P.; Gong, C.; Gillespie, M.; Griss, J.; Haw, R.; Jassal, B.; Matthews, L.; May, B.; et al. The Reactome Pathway Knowledgebase 2024. Nucleic Acids Res. 2024, 52, D672–D678. [Google Scholar] [CrossRef]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Blaser, S.; Schaye, V.; Hwang, J.; Cocks, P.; Kudlowitz, D. Hickam’s dictum, Occam’s razor, and Crabtree’s bludgeon: A case of renal failure and a clavicular mass. Diagnosis 2022, 9, 133–139. [Google Scholar] [CrossRef]
- Linscott, K.B.; Cassady, J.A.; Robin, N.H. Occam’s razor dulled: The occurrence of multiple genetic diagnoses. Curr. Opin. Pediatr. 2021, 33, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Spedicati, B.; Morgan, A.; Pianigiani, G.; Musante, L.; Rubinato, E.; Santin, A.; Nardone, G.G.; Faletra, F.; Girotto, G. Challenging Occam’s Razor: Dual Molecular Diagnoses Explain Entangled Clinical Pictures. Genes 2022, 13, 2023. [Google Scholar] [CrossRef] [PubMed]
- Katsanis, N. The continuum of causality in human genetic disorders. Genome Biol. 2016, 17, 233. [Google Scholar] [CrossRef] [PubMed]
- Rosina, E.; Pezzani, L.; Pezzoli, L.; Marchetti, D.; Bellini, M.; Pilotta, A.; Calabrese, O.; Nicastro, E.; Cirillo, F.; Cereda, A.; et al. Atypical, Composite, or Blended Phenotypes: How Different Molecular Mechanisms Could Associate in Double-Diagnosed Patients. Genes 2022, 13, 1275. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, S.; Gravel, B.; Nachtegael, C.; De Baere, E.; Loeys, B.; Vikkula, M.; Smits, G.; Lenaerts, T. Toward reporting standards for the pathogenicity of variant combinations involved in multilocus/oligogenic diseases. Hum. Genet. Genom. Adv. 2023, 4, 100165. [Google Scholar] [CrossRef] [PubMed]
- Ameratunga, R.; Woon, S.-T.; Bryant, V.L.; Steele, R.; Slade, C.; Leung, E.Y.; Lehnert, K. Clinical Implications of Digenic Inheritance and Epistasis in Primary Immunodeficiency Disorders. Front. Immunol. 2017, 8, 1965. [Google Scholar] [CrossRef]
- Suls, A.; Velizarova, R.; Yordanova, I.; Deprez, L.; Van Dyck, T.; Wauters, J.; Guergueltcheva, V.; Claes, L.; Kremensky, I.; Jordanova, A.; et al. Four generations of epilepsy caused by an inherited microdeletion of the SCN1A gene. Neurology 2010, 75, 72–76. [Google Scholar] [CrossRef]
- Depienne, C.; Trouillard, O.; Gourfinkel-An, I.; Saint-Martin, C.; Bouteiller, D.; Graber, D.; Barthez-Carpentier, M.-A.; Gautier, A.; Villeneuve, N.; Dravet, C.; et al. Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. J. Med. Genet. 2010, 47, 404–410. [Google Scholar] [CrossRef]
- de Lange, I.M.; Mulder, F.; van.’t Slot, R.; Sonsma, A.C.M.; van Kempen, M.J.A.; Nijman, I.J.; Ernst, R.F.; Knoers, N.V.A.M.; Brilstra, E.H.; Koeleman, B.P.C. Modifier genes in SCN1A-related epilepsy syndromes. Mol. Genet. Genom. Med. 2020, 8, e1103. [Google Scholar] [CrossRef]
- Calhoun, J.D.; Hawkins, N.A.; Zachwieja, N.J.; Kearney, J.A. Cacna1g is a genetic modifier of epilepsy in a mouse model of Dravet syndrome. Epilepsia 2017, 58, e111–e115. [Google Scholar] [CrossRef]
- Hawkins, N.A.; Kearney, J.A. Hlf is a genetic modifier of epilepsy caused by voltage-gated sodium channel mutations. Epilepsy Res. 2016, 119, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Dash, B.; Han, C.; Waxman, S.G.; Dib-Hajj, S.D. Nonmuscle myosin II isoforms interact with sodium channel α subunits. Mol. Pain 2018, 14, 1744806918788638. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Adelstein, R.S. The role of vertebrate nonmuscle Myosin II in development and human disease. BioArchitecture 2014, 4, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Newell-Litwa, K.A.; Horwitz, R.; Lamers, M.L. Non-muscle myosin II in disease: Mechanisms and therapeutic opportunities. Dis. Model. Mech. 2015, 8, 1495–1515. [Google Scholar] [CrossRef]
- Javier-Torrent, M.; Saura, C.A. Conventional and Non-Conventional Roles of Non-Muscle Myosin II-Actin in Neuronal Development and Degeneration. Cells 2020, 9, 1926. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Person | Gender | Onset Age (Years) | Type of Seizure | Age Range of Seizures (Years) | Fever/Temp (°C) | Duration of Longest Seizure | Duration of Episode/How Many Seizures Per Episode | Frequency of Episodes | On Treatment at the Time |
|---|---|---|---|---|---|---|---|---|---|
| Patient | M | 4 | Generalized tonic-clonic | 4~5 | Yes/39.8 | 10 min | 5 days/20+ times | Twice in one year | Levetiracetam |
| Mother | F | 28 | Generalized tonic-clonic | 28 | Yes/39.5 | 15 s | Once | – | Supportive treatment |
| Aunt | F | 5 | Generalized tonic-clonic | 5 | Yes/– | 10 s | Once | – | Supportive treatment |
| Cousin | F | 1 | Generalized tonic-clonic | 1 | Yes/– | 10 s | Once | – | Supportive treatment |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, S.; Niu, Z.; Qiao, X. Exploring the Genotype–Phenotype Correlations in a Child with Inherited Seizure and Thrombocytopenia by Digenic Network Analysis. Genes 2024, 15, 1004. https://doi.org/10.3390/genes15081004
Lu S, Niu Z, Qiao X. Exploring the Genotype–Phenotype Correlations in a Child with Inherited Seizure and Thrombocytopenia by Digenic Network Analysis. Genes. 2024; 15(8):1004. https://doi.org/10.3390/genes15081004
Chicago/Turabian StyleLu, Shuanglong, Zhixiao Niu, and Xiaohong Qiao. 2024. "Exploring the Genotype–Phenotype Correlations in a Child with Inherited Seizure and Thrombocytopenia by Digenic Network Analysis" Genes 15, no. 8: 1004. https://doi.org/10.3390/genes15081004