

A Novel Pathogenic TUBA1A Variant in a Croatian Infant Is Linked to a Severe Tubulinopathy with Walker–Warburg-like Features

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Investigation
2.2. Genetic Investigation
2.3. Analysis of Mutation Location in the 3D Structure of Tubulin α-1A Chain Protein in a Complex with Tubulin β-III Protein
3. Results
3.1. Clinical Investigations
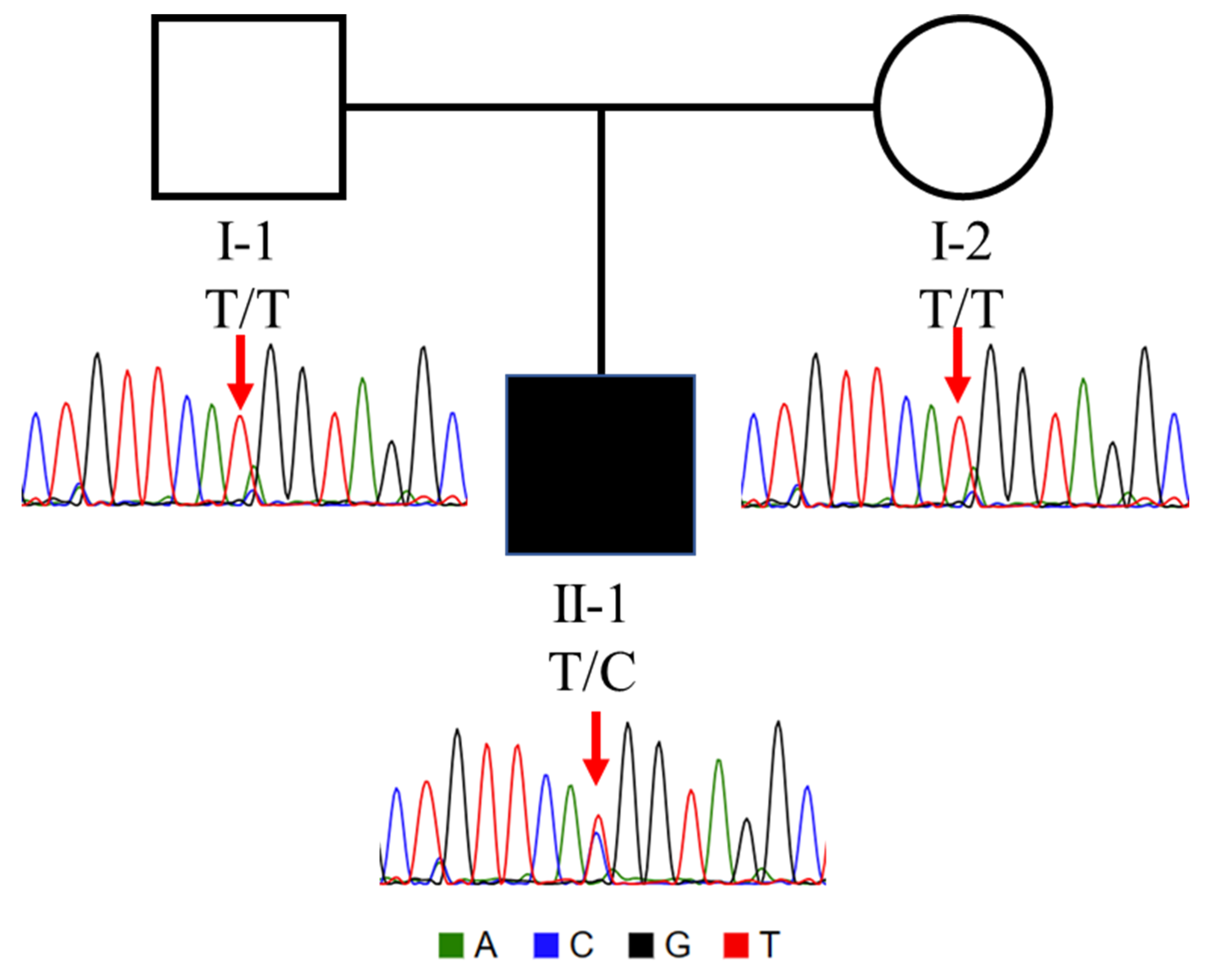
3.2. Genetic Investigation
3.3. Analysis of Mutation Location in the 3D Structure of Tubulin α-1A Chain Protein in a Complex with β-Tubulin
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hebebrand, M.; Huffmeier, U.; Trollmann, R.; Hehr, U.; Uebe, S.; Ekici, A.B.; Kraus, C.; Krumbiegel, M.; Reis, A.; Thiel, C.T.; et al. The mutational and phenotypic spectrum of tuba1a-associated tubulinopathy. Orphanet J. Rare Dis. 2019, 14, 38. [Google Scholar] [CrossRef] [PubMed]
- Buscaglia, G.; Northington, K.R.; Aiken, J.; Hoff, K.J.; Bates, E.A. Bridging the gap: The importance of tuba1a α-tubulin in forming midline commissures. Front. Cell Dev. Biol. 2021, 9, 789438. [Google Scholar] [CrossRef] [PubMed]
- Dobyns, W.B.; Pagon, R.A.; Armstrong, D.; Curry, C.J.; Greenberg, F.; Grix, A.; Holmes, L.B.; Laxova, R.; Michels, V.V.; Robinow, M.; et al. Diagnostic criteria for walker-warburg syndrome. Am. J. Med. Genet. 1989, 32, 195–210. [Google Scholar] [CrossRef]
- Vajsar, J.; Schachter, H. Walker-warburg syndrome. Orphanet J. Rare Dis. 2006, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Valero de Bernabe, D.; Currier, S.; Steinbrecher, A.; Celli, J.; van Beusekom, E.; van der Zwaag, B.; Kayserili, H.; Merlini, L.; Chitayat, D.; Dobyns, W.B.; et al. Mutations in the o-mannosyltransferase gene pomt1 give rise to the severe neuronal migration disorder walker-warburg syndrome. Am. J. Hum. Genet. 2002, 71, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Valero de Bernabe, D.; Voit, T.; Longman, C.; Steinbrecher, A.; Straub, V.; Yuva, Y.; Herrmann, R.; Sperner, J.; Korenke, C.; Diesen, C.; et al. Mutations in the fkrp gene can cause muscle-eye-brain disease and walker-warburg syndrome. J. Med. Genet. 2004, 41, e61. [Google Scholar] [CrossRef] [PubMed]
- Riemersma, M.; Mandel, H.; van Beusekom, E.; Gazzoli, I.; Roscioli, T.; Eran, A.; Gershoni-Baruch, R.; Gershoni, M.; Pietrokovski, S.; Vissers, L.E.; et al. Absence of α- and β-dystroglycan is associated with walker-warburg syndrome. Neurology 2015, 84, 2177–2182. [Google Scholar] [CrossRef]
- Hercus, C. Novoalign v4.4. Novocraft Technologies. Linux. 2022. Available online: https://www.novocraft.com/support/download/ (accessed on 27 July 2024).
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From fastq data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 10–11. [Google Scholar] [CrossRef]
- Cingolani, P.; Patel, V.M.; Coon, M.; Nguyen, T.; Land, S.J.; Ruden, D.M.; Lu, X. Using drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, snpsift. Front. Genet. 2012, 3, 35. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, snpeff: Snps in the genome of drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Liu, X.; Li, C.; Mou, C.; Dong, Y.; Tu, Y. Dbnsfp v4: A comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site snvs. Genome Med. 2020, 12, 103. [Google Scholar] [CrossRef]
- Pedersen, B.S.; Brown, J.M.; Dashnow, H.; Wallace, A.D.; Velinder, M.; Tristani-Firouzi, M.; Schiffman, J.D.; Tvrdik, T.; Mao, R.; Best, D.H.; et al. Effective variant filtering and expected candidate variant yield in studies of rare human disease. NPJ Genom. Med. 2021, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, K. Intervar: Clinical interpretation of genetic variants by the 2015 acmg-amp guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Jay, J.J.; Brouwer, C. Lollipops in the clinic: Information dense mutation plots for precision medicine. PLoS ONE 2016, 11, e0160519. [Google Scholar] [CrossRef] [PubMed]
- Tantry, M.S.A.; Santhakumar, K. Insights on the role of α- and β-tubulin isotypes in early brain development. Mol. Neurobiol. 2023, 60, 3803–3823. [Google Scholar] [CrossRef]
- Schrodinger, LLC. The Pymol Molecular Graphics System, version 1.8; Schrodinger, LLC.: New York, NY, USA, 2015. [Google Scholar]
- Vemu, A.; Atherton, J.; Spector, J.O.; Szyk, A.; Moores, C.A.; Roll-Mecak, A. Structure and dynamics of single-isoform recombinant neuronal human tubulin. J. Biol. Chem. 2016, 291, 12907–12915. [Google Scholar] [CrossRef] [PubMed]
- Voigt, M.; Rochow, N.; Schneider, K.T.; Hagenah, H.P.; Scholz, R.; Hesse, V.; Wittwer-Backofen, U.; Straube, S.; Olbertz, D. New percentile values for the anthropometric dimensions of singleton neonates: Analysis of perinatal survey data of 2007-2011 from all 16 states of germany. Z. Geburtshilfe Neonatol. 2014, 218, 210–217. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Yokoi, S.; Ishihara, N.; Miya, F.; Tsutsumi, M.; Yanagihara, I.; Fujita, N.; Yamamoto, H.; Kato, M.; Okamoto, N.; Tsunoda, T.; et al. Tuba1a mutation can cause a hydranencephaly-like severe form of cortical dysgenesis. Sci. Rep. 2015, 5, 15165. [Google Scholar] [CrossRef]
- Zocchi, R.; Bellacchio, E.; Piccione, M.; Scardigli, R.; D’Oria, V.; Petrini, S.; Baranano, K.; Bertini, E.; Sferra, A. Novel loss of function mutation in tuba1a gene compromises tubulin stability and proteostasis causing spastic paraplegia and ataxia. Front. Cell. Neurosci. 2023, 17, 1162363. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Francioli, L.C.; Goodrich, J.K.; Collins, R.L.; Kanai, M.; Wang, Q.; Alfoldi, J.; Watts, N.A.; Vittal, C.; Gauthier, L.D.; et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 2024, 625, 92–100. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Pedigree Number | II-1 |
| Sex | Male |
| Ethnicity | Croatian |
| Parental consanguinity | Non-consanguineous |
| Clinical Features | |
| Age at onset | Birth |
| Current age or age of death | Death at 2 months of age |
| Developmental delay or intellectual impairment | N/A |
| Gait abnormalities | N/A |
| Ataxia | N/A |
| Seizures | + |
| Neuropathy | N/A |
| Macrocephaly | + |
| Hypoplastic genitalia, micropenis | + |
| cMRI Findings (affected regions) | |
| Basal ganglia | + |
| Cortex | + |
| Corpus callosum | + |
| Cerebellum | + |
| Source/Database | ClinVar | Tantry et al. (2023) [16] | Case |
|---|---|---|---|
| Number of Mutations/Variations reported | 46 | 24 | 1 |
| Phenotypes | |||
| Lissencephaly | + | + | + |
| Seizures/Epilepsy | + | + | + |
| Brain malformations (Cortex, Brain Stem) | + | ||
| 4th ventricle dilatation/Dandy Walker malformation/variant | + | ||
| Absence of language | + | ||
| Anomalous structures of the brainstem | + | ||
| Autism | + | ||
| Cerebellar hypoplasia | + | + | |
| Congenital cataracts | + | ||
| Corpus callosum agenesis | + | ||
| Hypotonia | + | ||
| Intellectual disability | + | ||
| Macrocephaly | + | ||
| Microcephaly | + | ||
| Microphthalmia | + | ||
| Pachygyria | + | ||
| Polymicrogyria | + | ||
| Polymicrogyria-like | + | ||
| Rudimentary and unconnected basal ganglia | + | ||
| Ventricular dilatation | + | ||
| Vermian aplasia (Cerebellar vermis aplasia) | + | ||
| Hypoplastic genitalia + micropenis | + |
| TUBA1A Tubulinopathy | Patient | WWS |
|---|---|---|
| Microlysencephaly | ||
| Lysencephaly | Lysencephaly | Lysencephaly |
| Polimicrogyria-like cortical dysplasia | ||
| Corpus callosum agenesis | Corpus callosum agenesis | Corpus callosum hypoplasia/agenesis |
| Disorganized stiatum and thalami | Rudimentary and unconnected basal ganglia | |
| Cerebellar hypoplasia | Cerebellar hypoplasia | Cerebellar hypoplasia |
| Vermian hypoplasia | Vermian aplasia | Vermian hypoplasia |
| Brainstem hypoplasia | Anomalous structures of the brainstem | Flat brainstem |
| Ventricular dilatation | Ventricular dilatation | Ventricular dilatation |
| 4th ventricle dilatation | 4th ventricle dilatation/Dandy Walker malformation/variant | Dandy Walker malformation |
| Microcephaly | Macrocephaly | |
| Developmental delay | N/A | Developmental delay |
| Intellectual disability | N/A | Mental retardation |
| Seizures | Seizures | Seizures |
| Hypotonia | Hypotonia | Hypotonia |
| Eye malformations | Eye malformations | |
| Hypoplastic genitalia + micropenis | Undescended testes + micropenis | |
| Muscle weakness/congenital muscular dystrophy | ||
| White matter hypomyelination | ||
| Facial dysmorphic features |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saidin, A.; Papazovska Cherepnalkovski, A.; Shaukat, Z.; Arsov, T.; Hussain, R.; Roberts, B.J.; Bucat, M.; Cogelja, K.; Ricos, M.G.; Dibbens, L.M. A Novel Pathogenic TUBA1A Variant in a Croatian Infant Is Linked to a Severe Tubulinopathy with Walker–Warburg-like Features. Genes 2024, 15, 1031. https://doi.org/10.3390/genes15081031
Saidin A, Papazovska Cherepnalkovski A, Shaukat Z, Arsov T, Hussain R, Roberts BJ, Bucat M, Cogelja K, Ricos MG, Dibbens LM. A Novel Pathogenic TUBA1A Variant in a Croatian Infant Is Linked to a Severe Tubulinopathy with Walker–Warburg-like Features. Genes. 2024; 15(8):1031. https://doi.org/10.3390/genes15081031
Chicago/Turabian StyleSaidin, Akzam, Anet Papazovska Cherepnalkovski, Zeeshan Shaukat, Todor Arsov, Rashid Hussain, Ben J. Roberts, Marija Bucat, Klara Cogelja, Michael G. Ricos, and Leanne M. Dibbens. 2024. "A Novel Pathogenic TUBA1A Variant in a Croatian Infant Is Linked to a Severe Tubulinopathy with Walker–Warburg-like Features" Genes 15, no. 8: 1031. https://doi.org/10.3390/genes15081031
APA StyleSaidin, A., Papazovska Cherepnalkovski, A., Shaukat, Z., Arsov, T., Hussain, R., Roberts, B. J., Bucat, M., Cogelja, K., Ricos, M. G., & Dibbens, L. M. (2024). A Novel Pathogenic TUBA1A Variant in a Croatian Infant Is Linked to a Severe Tubulinopathy with Walker–Warburg-like Features. Genes, 15(8), 1031. https://doi.org/10.3390/genes15081031