
A Potential Role for the Amyloid Precursor Protein in the Regulation of Interferon Signaling, Cholesterol Homeostasis, and Tau Phosphorylation in Niemann–Pick Disease Type C
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Transcriptome Analysis
2.3. Data Preprocessing
2.4. Differential Gene Expression Analysis
2.5. Ingenuity Pathway Analysis
2.6. Gene Set Enrichment Analysis
3. Results
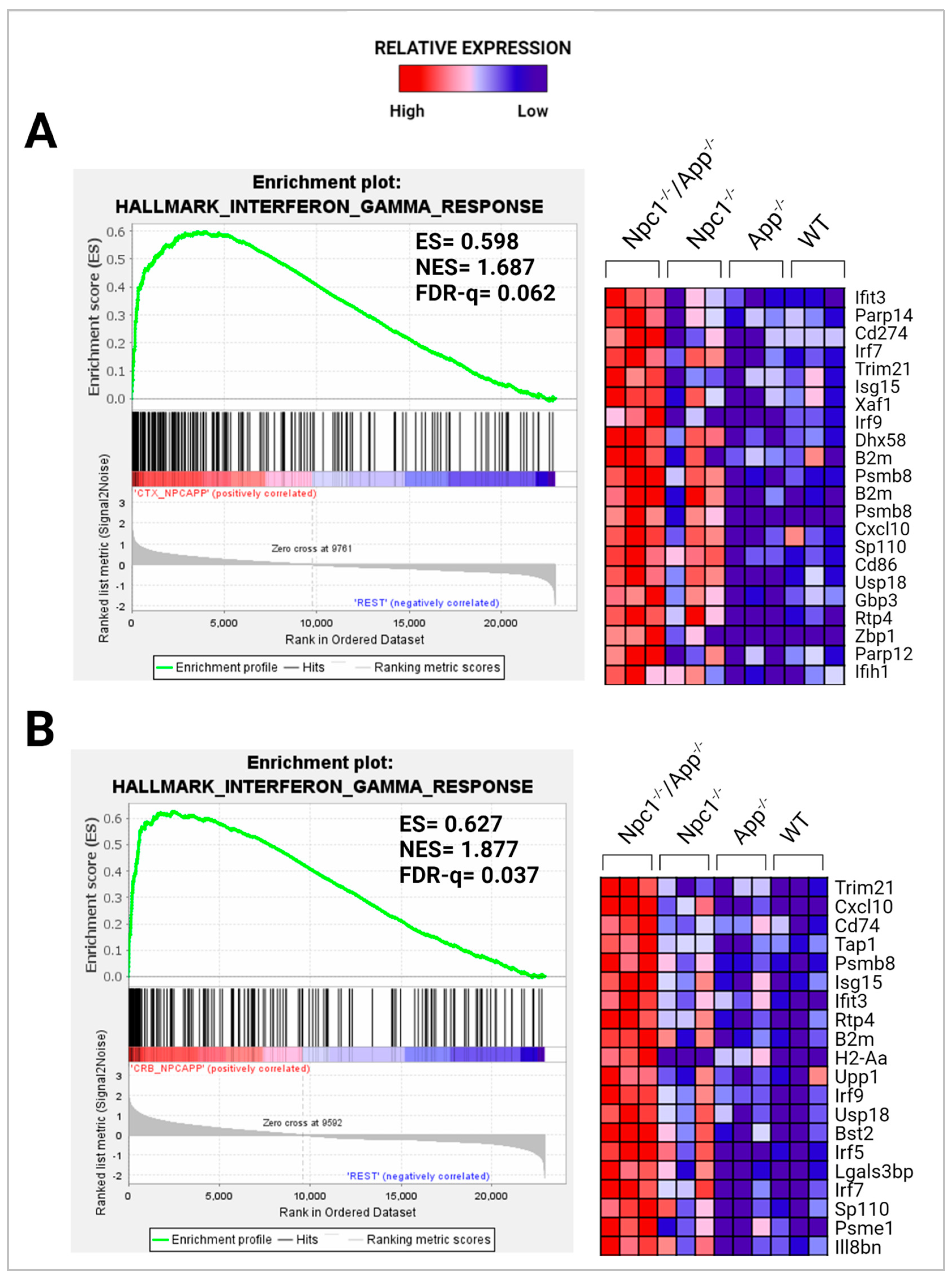
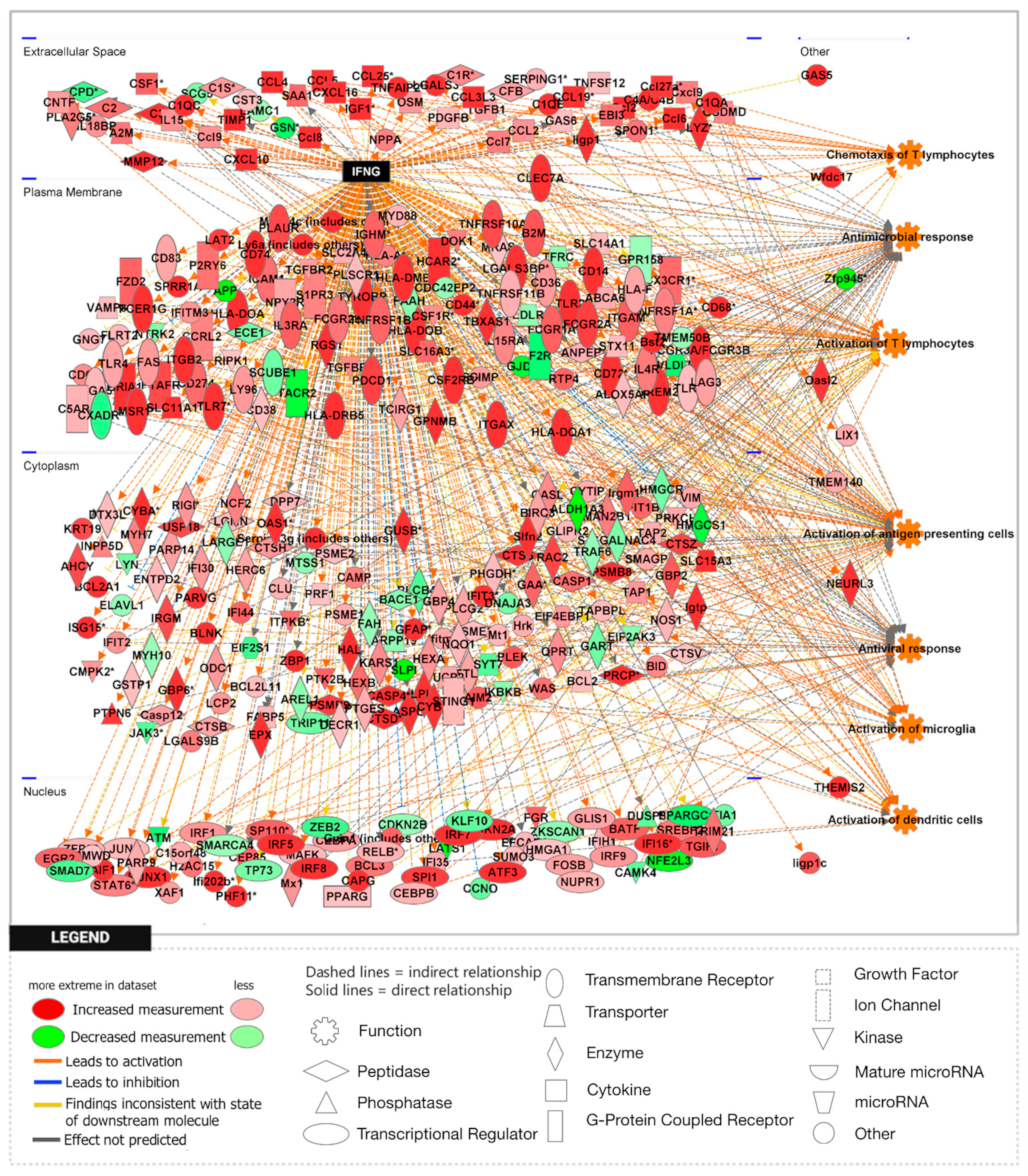
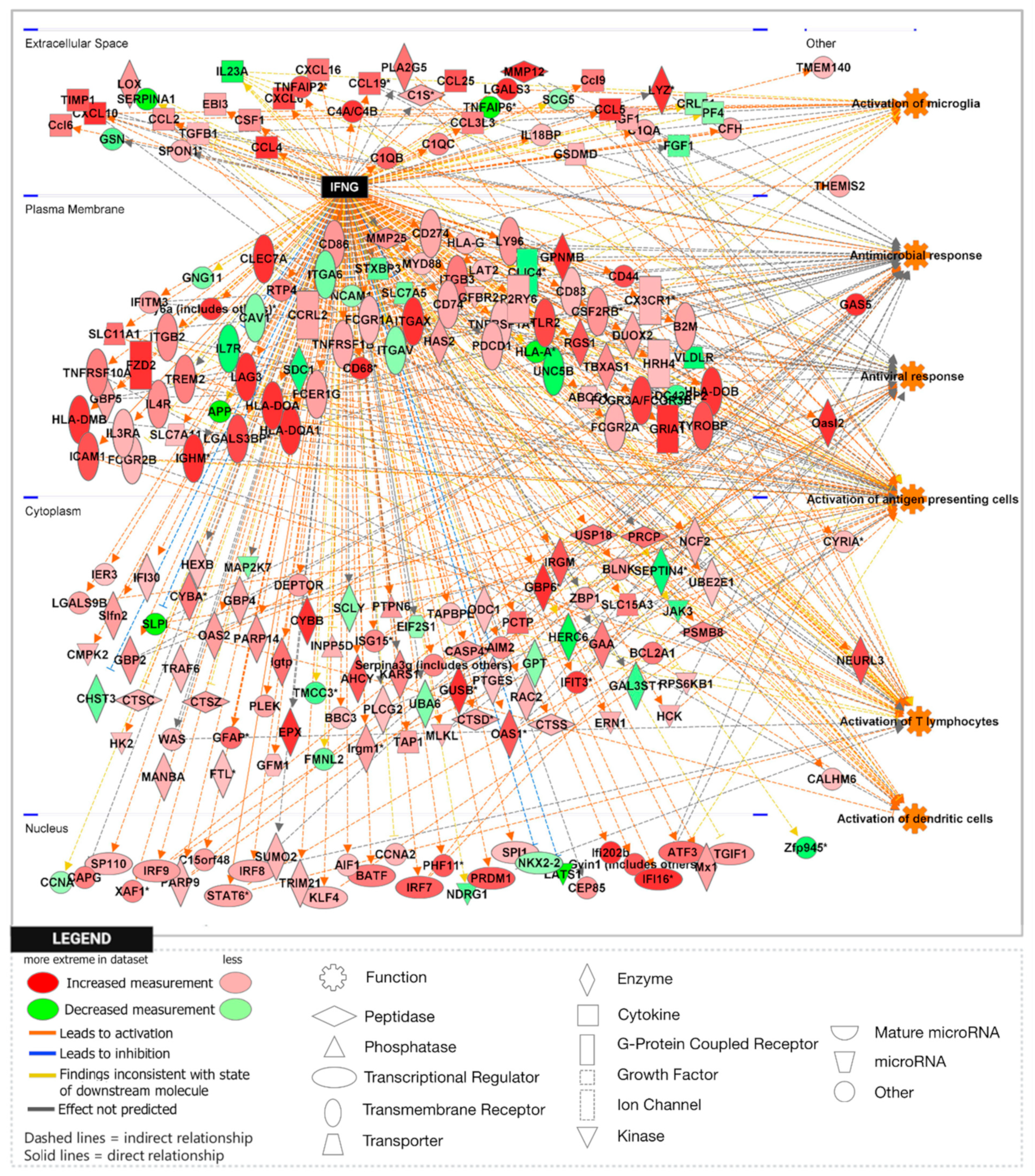
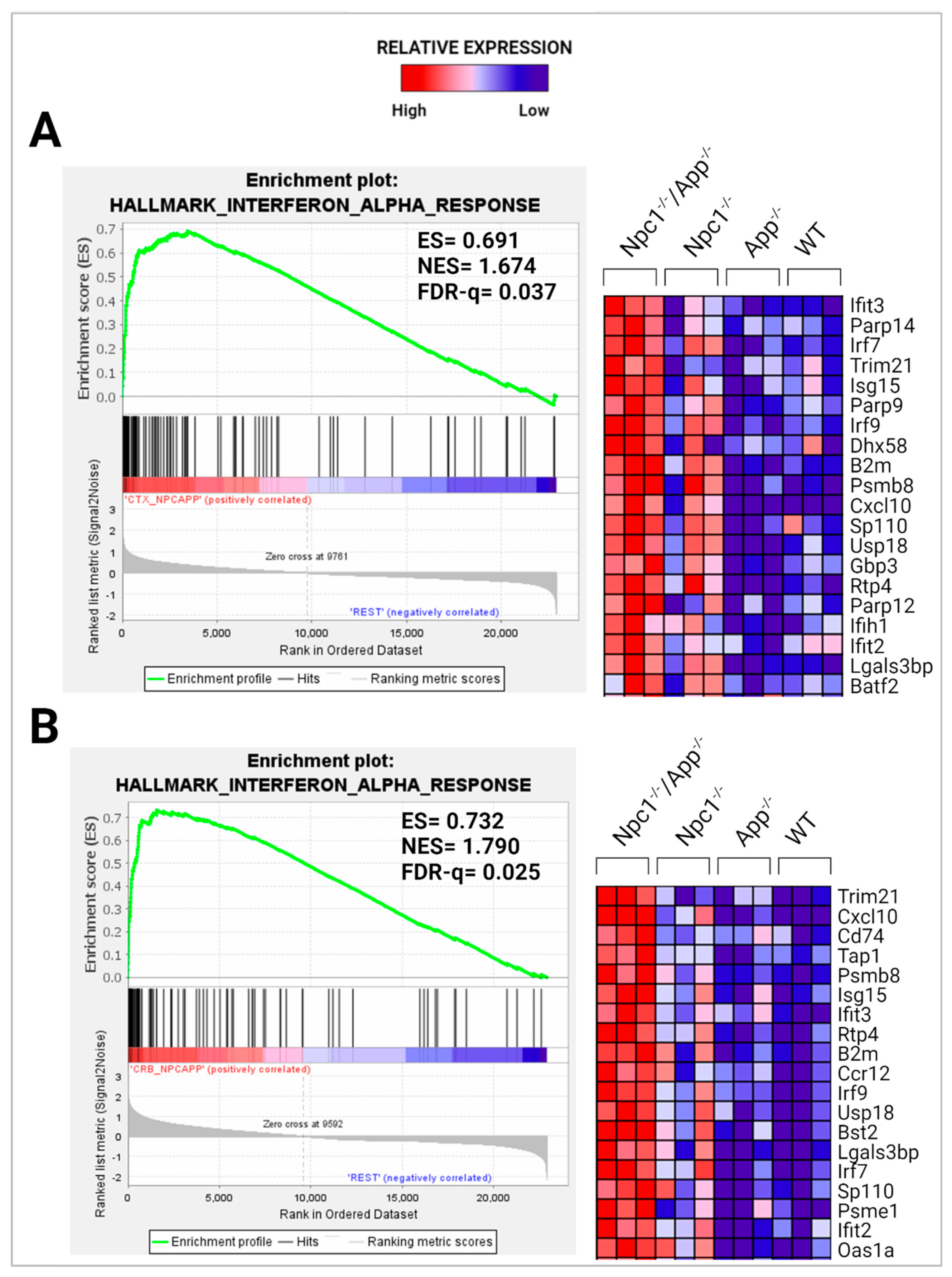
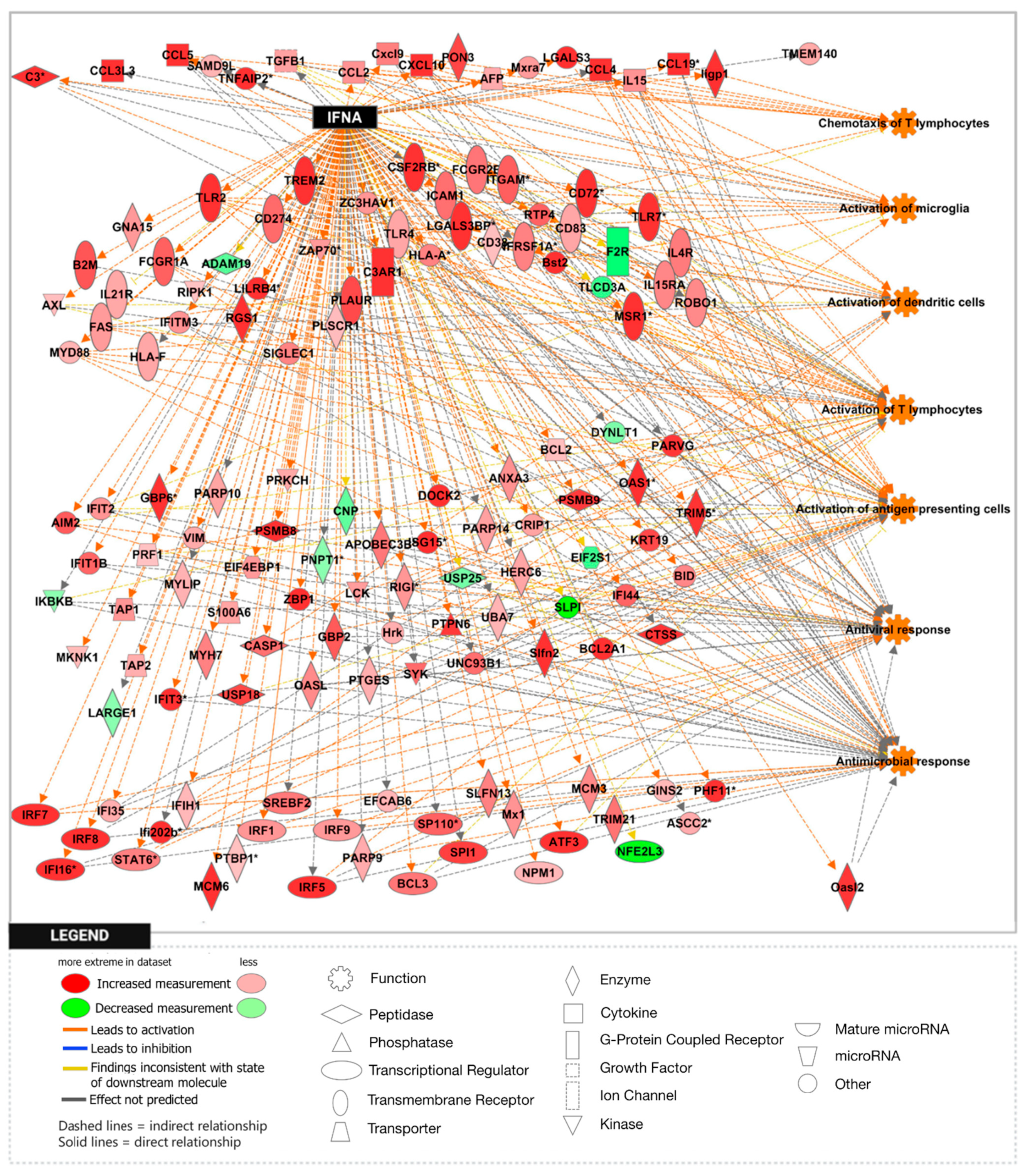
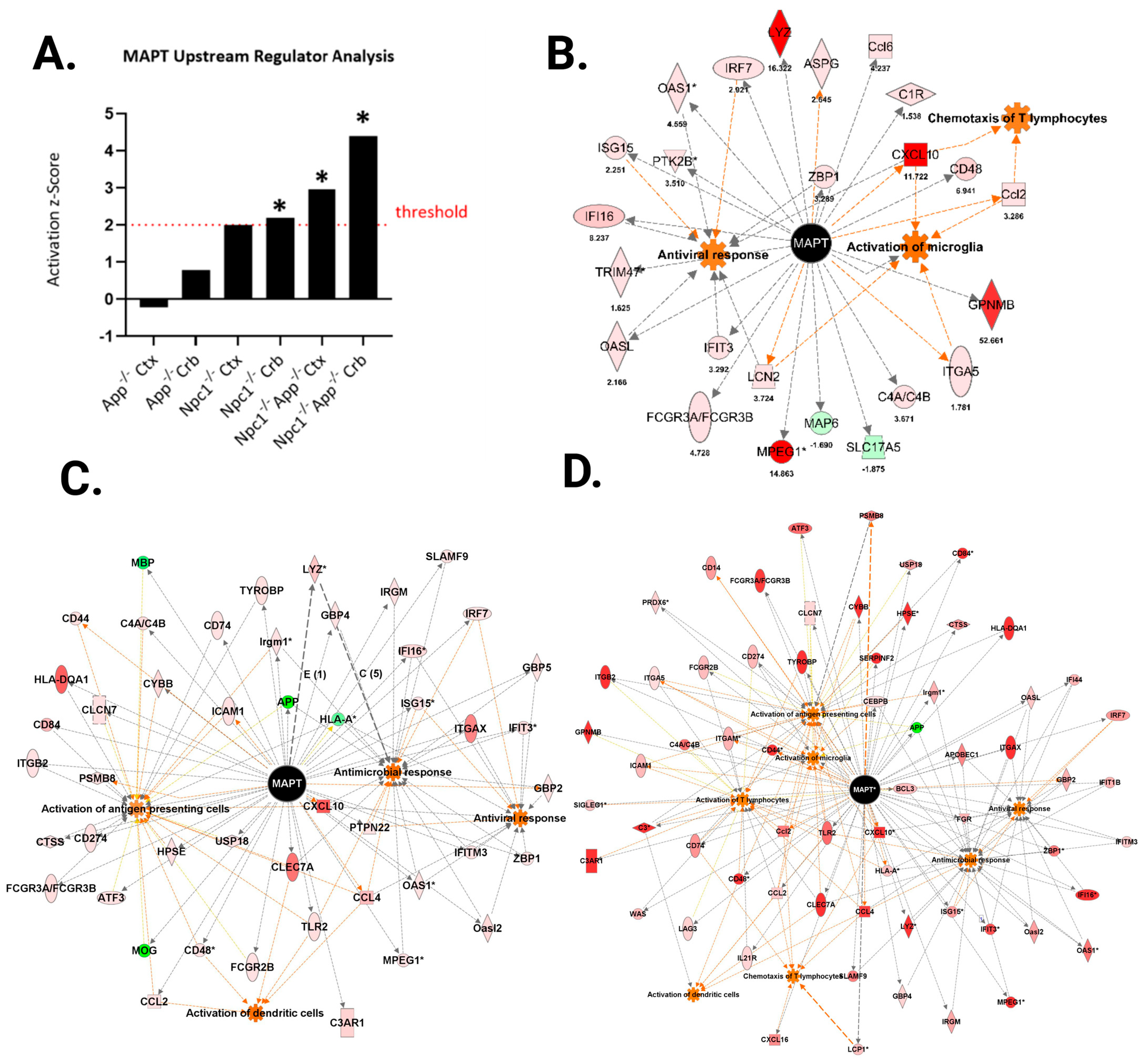
3.1. Loss of APP Amplifies Interferon Signaling in the Npc1−/− Cerebral Cortex and Cerebellum
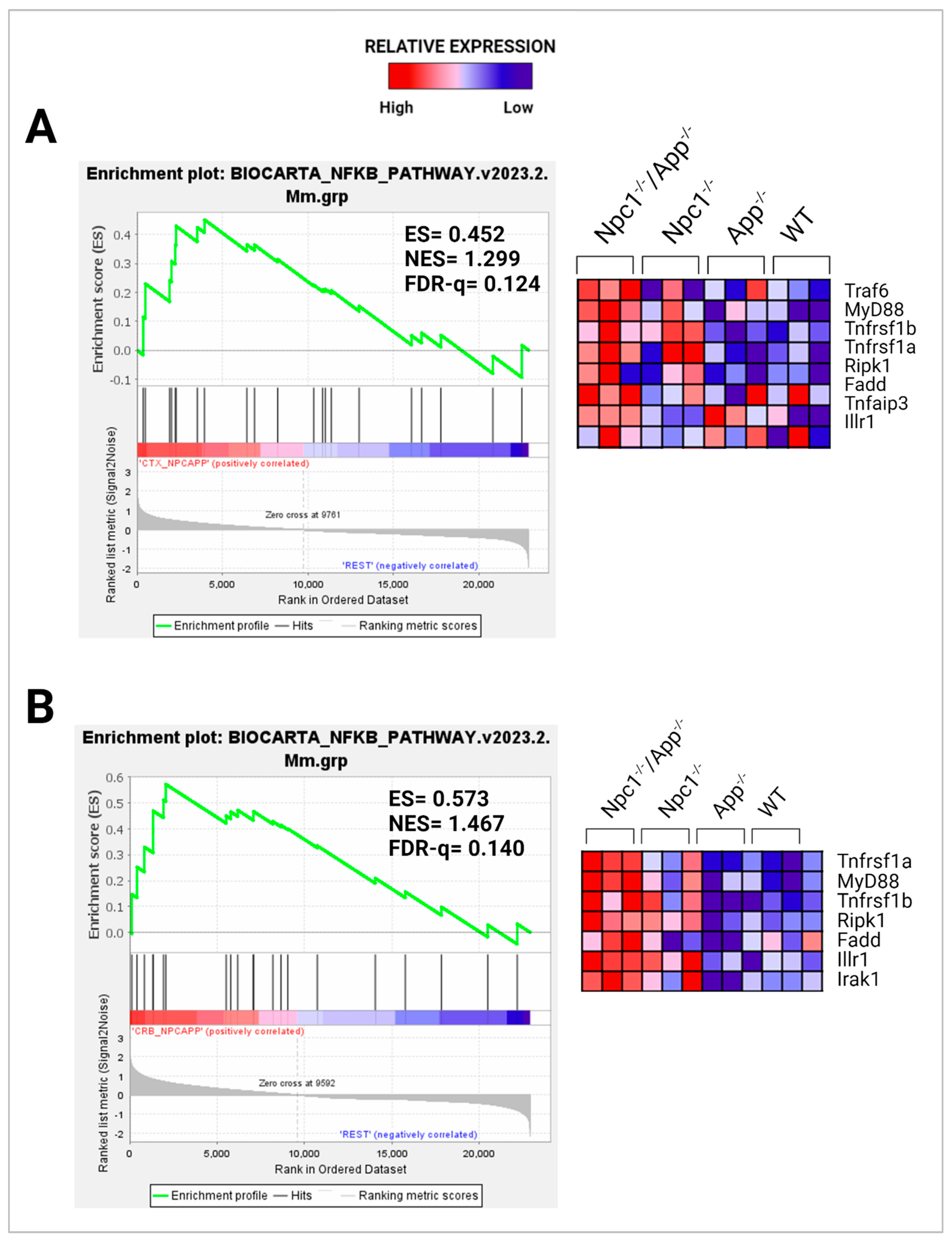
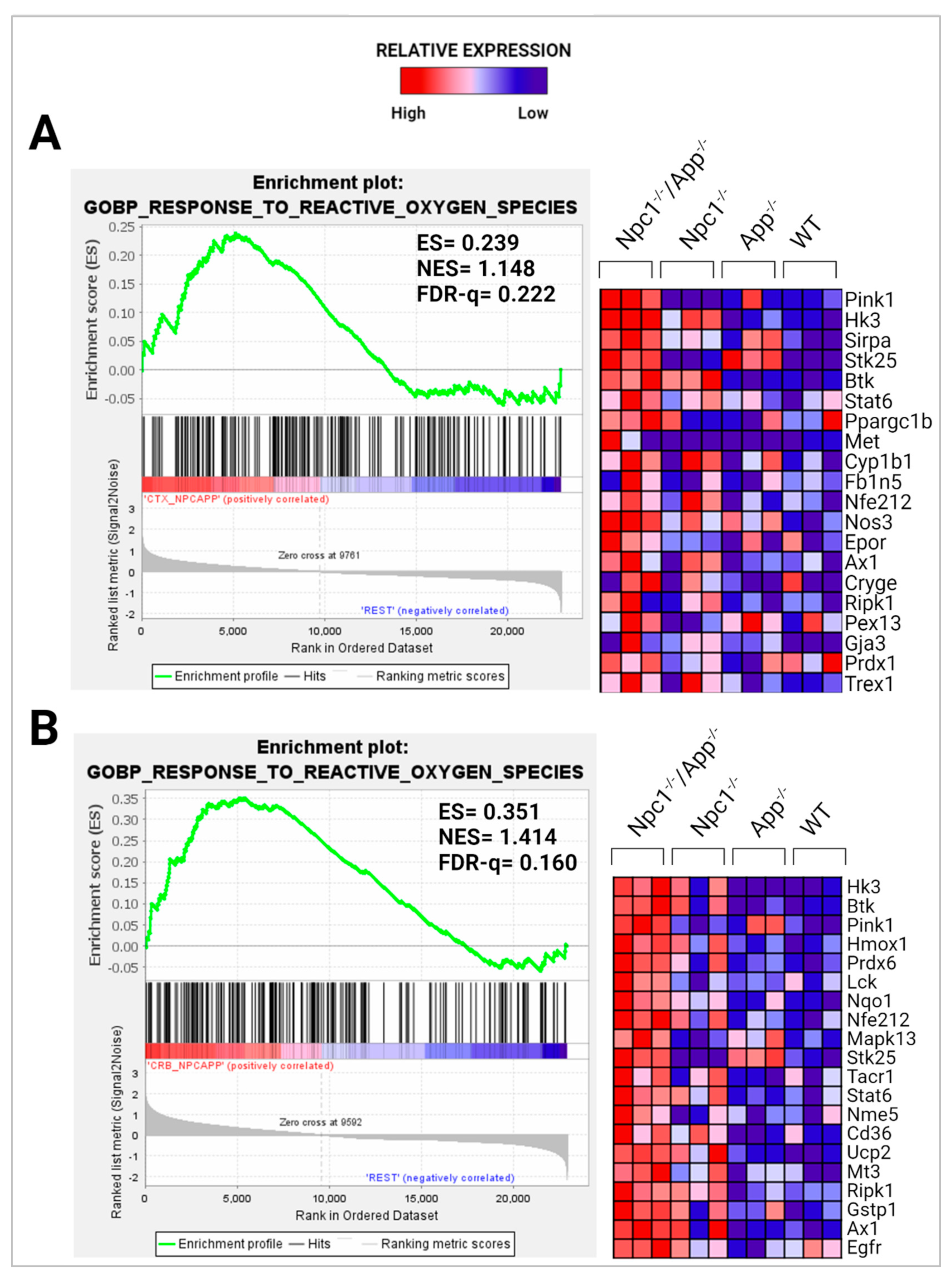
3.2. APP Depletion Exacerbates Inflammatory Pathways Linked To DAMP Generation, ROS, and Lipid Peroxidation in the Npc1−/− Cerebral Cortex
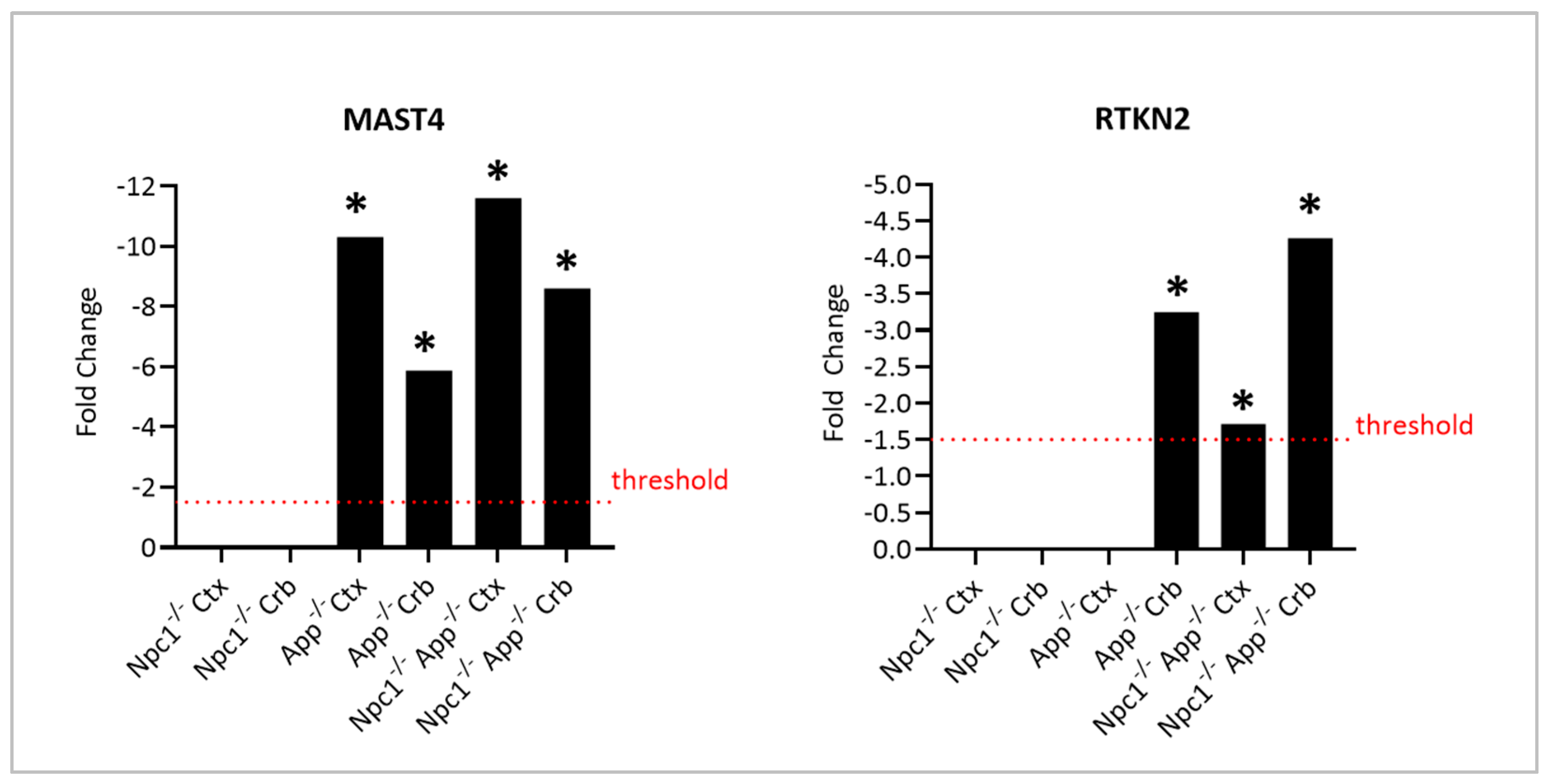
3.3. The Cellular Hormetic Response to 27-Hydroxycholesterol Is Impaired Following APP Depletion in the NPC Brain
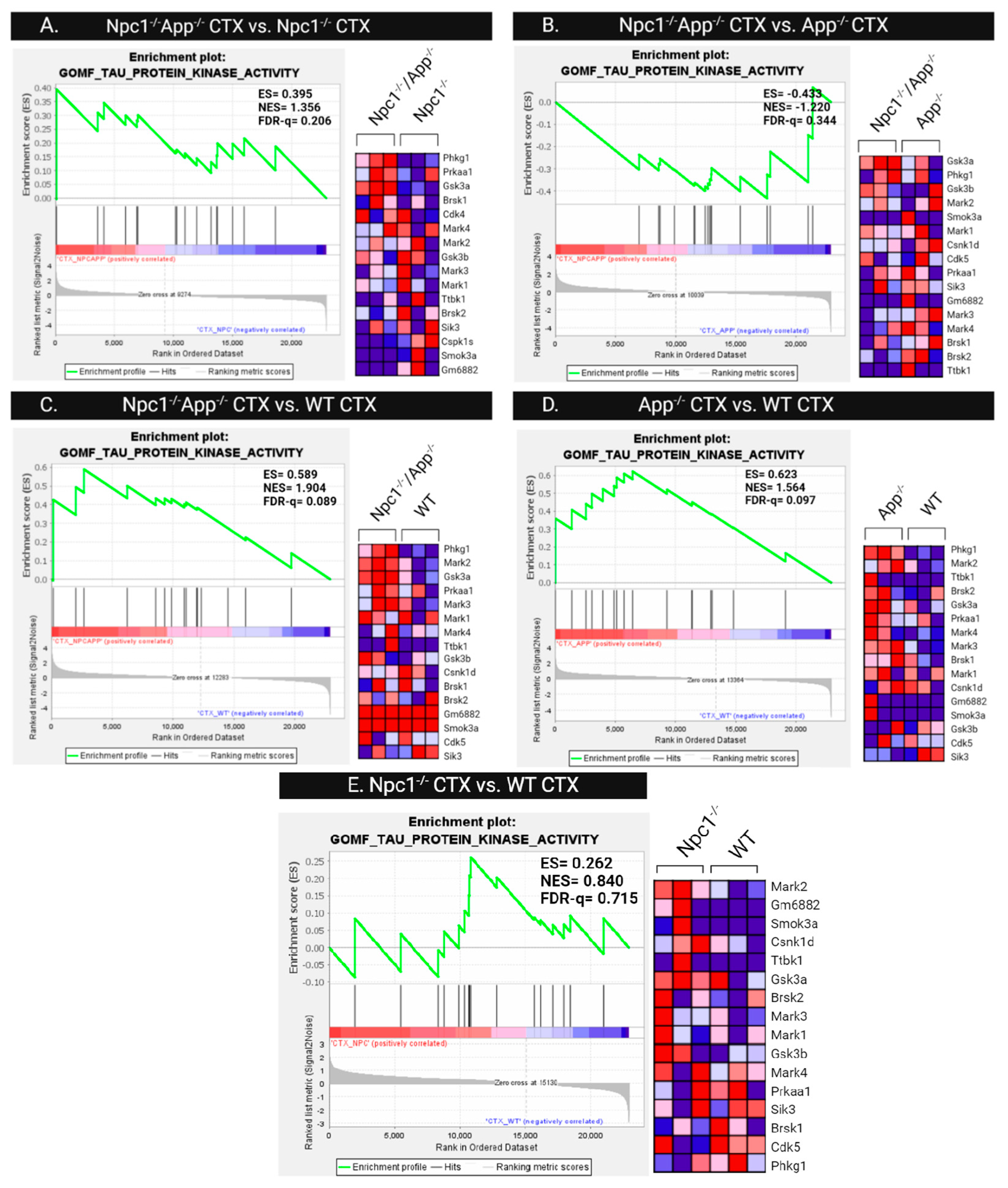
3.4. Npc1−/− Tau Hyperphosphorylation Is Exacerbated by APP Depletion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Burton, B.K.; Ellis, A.G.; Orr, B.; Chatlani, S.; Yoon, K.; Shoaff, J.R.; Gallo, D. Estimating the prevalence of Niemann-Pick disease type C (NPC) in the United States. Mol. Genet. Metab. 2021, 134, 182–187. [Google Scholar] [CrossRef]
- Yanjanin, N.M.; Vélez, J.I.; Gropman, A.; King, K.; Bianconi, S.E.; Conley, S.K.; Brewer, C.C.; Solomon, B.; Pavan, W.J.; Arcos-Burgos, M.; et al. Linear clinical progression, independent of age of onset, in Niemann–Pick disease, type C. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153B, 132–140. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef]
- Shin, S.D.; Shin, A.; Mayagoitia, K.; Wilson, C.G.; Bellinger, D.L.; Soriano, S. Interferon downstream signaling is activated early in pre-symptomatic Niemann-Pick disease type C. Neurosci. Lett. 2019, 706, 43–50. [Google Scholar] [CrossRef]
- Shin, S.D.; Shin, A.; Mayagoitia, K.; Siebold, L.; Rubini, M.; Wilson, C.G.; Bellinger, D.L.; Soriano, S. Loss of amyloid precursor protein exacerbates early inflammation in Niemann-Pick disease type C. J. Neuroinflammation 2019, 16, 269. [Google Scholar] [CrossRef]
- Ong, W.Y.; Kumar, U.; Switzer, R.C.; Sidhu, A.; Suresh, G.; Hu, C.Y.; Patel, S.C. Neurodegeneration in Niemann-Pick type C disease mice. Exp. Brain Res. 2001, 141, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, H.; Azhar, W. Niemann-Pick Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK556129/ (accessed on 13 June 2024).
- Mengel, E.; Klünemann, H.H.; Lourenço, C.M.; Hendriksz, C.J.; Sedel, F.; Walterfang, M.; Kolb, S.A. Niemann-Pick disease type C symptomatology: An expert-based clinical description. Orphanet J. Rare Dis. 2013, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.; Pressey, S.N.R.; Cooper, J.D.; Soriano, S. Loss of amyloid precursor protein in a mouse model of Niemann–Pick type C disease exacerbates its phenotype and disrupts tau homeostasis. Neurobiol. Dis. 2011, 42, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Kodam, A.; Maulik, M.; Peake, K.; Amritraj, A.; Vetrivel, K.S.; Thinakaran, G.; Vance, J.E.; Kar, S. Altered levels and distribution of APP and its processing enzymes in Niemann-Pick Type C1-deficient mouse brains. Glia 2010, 58, 1267–1281. [Google Scholar] [CrossRef]
- Tolan, A.J.; Sanchez, K.L.; Shin, S.D.; White, J.B.; Currais, A.; Soriano-Castell, D.; Wilson, C.G.; Maher, P.; Soriano, S. Differential Interferon Signaling Regulation and Oxidative Stress Responses in the Cerebral Cortex and Cerebellum Could Account for the Spatiotemporal Pattern of Neurodegeneration in Niemann–Pick Disease Type C. Genes 2024, 15, 101. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Mellon, S.H.; Butters, T.D.; Nevyjel, M.; Covey, D.F.; Bembi, B.; Dardis, A. Oxidative stress in Npc1 deficient cells: Protective effect of allopregnanolone. J. Cell. Mol. Med. 2009, 13, 3786–3796. [Google Scholar] [CrossRef]
- Kielian, T. Lysosomal storage disorders: Pathology within the lysosome and beyond. J. Neurochem. 2019, 148, 568–572. Available online: https://onlinelibrary.wiley.com/doi/10.1111/jnc.14672 (accessed on 22 May 2024). [CrossRef] [PubMed]
- Suzuki, M.; Sugimoto, Y.; Ohsaki, Y.; Ueno, M.; Kato, S.; Kitamura, Y.; Hosokawa, H.; Davies, J.P.; Ioannou, Y.A.; Vanier, M.T.; et al. Endosomal Accumulation of Toll-Like Receptor 4 Causes Constitutive Secretion of Cytokines and Activation of Signal Transducers and Activators of Transcription in Niemann–Pick Disease Type C (NPC) Fibroblasts: A Potential Basis for Glial Cell Activation in the NPC Brain. J. Neurosci. 2007, 27, 1879–1891. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Sig. Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.-T.; Tu, X.; Yang, K.; Wu, J.; Repa, J.J.; Yan, N. Tonic prime-boost of STING signaling mediates Niemann–Pick disease type C. Nature 2021, 596, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Gongol, B.; Marin, T.L.; Jeppson, J.D.; Mayagoitia, K.; Shin, S.; Sanchez, N.; Kirsch, W.M.; Vinters, H.V.; Wilson, C.G.; Ghribi, O.; et al. Cellular hormetic response to 27-hydroxycholesterol promotes neuroprotection through AICD induction of MAST4 abundance and kinase activity. Sci. Rep. 2017, 7, 13898. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.-X.; Iqbal, K. Hyperphosphorylation of Microtubule-Associated Protein Tau: A Promising Therapeutic Target for Alzheimer Disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef]
- Caillet-Boudin, M.-L.; Buée, L.; Sergeant, N.; Lefebvre, B. Regulation of human MAPT gene expression. Mol. Neurodegener. 2015, 10, 28. [Google Scholar] [CrossRef]
- Winchester, J.S.; Rouchka, E.C.; Rowland, N.S.; Rice, N.A. In Silico characterization of phosphorylase kinase: Evidence for an alternate intronic polyadenylation site in PHKG1. Mol. Genet. Metab. 2007, 92, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Maruko-Otake, A.; Ohtake, Y.; Hayashishita, M.; Sekiya, M.; Iijima, K.M. Stabilization of Microtubule-Unbound Tau via Tau Phosphorylation at Ser262/356 by Par-1/MARK Contributes to Augmentation of AD-Related Phosphorylation and Aβ42-Induced Tau Toxicity. PLoS Genet. 2016, 12, e1005917. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cortex | Cerebellum | |||||
|---|---|---|---|---|---|---|
| Npc1−/− | App−/− | Npc1−/− /App−/− | *Npc1−/− | App−/− | *Npc1−/− /App−/− | |
| DEG (aFC + p) | 321 | 843 | 1884 | 387 | 1065 | 1973 |
| DEG (up) | 199 | 459 | 1177 | 176 | 457 | 1265 |
| DEG (down) | 122 | 384 | 707 | 211 | 608 | 708 |
| Upstream Regulators | Npc1−/−/App−/− Ctx vs. WT Ctx | *Npc1−/− Ctx vs. WT Ctx | App−/− Ctx vs. WT Ctx | ||||||
|---|---|---|---|---|---|---|---|---|---|
| z-Score | −log(p) | #T.M | z-Score | −log(p) | #T.M | z-Score | −log(p) | #T.M | |
| IFN-γ | 8.494 | 33.070 | 201 | 4.104 | 10.291 | 84 | −0.844 | 3.650 | 66 |
| M-CSF/CSF1 | 4.257 | 41.633 | 116 | 3.785 | 5.210 | 21 | 0.588 | 1.527 | 16 |
| GM-CSF/CSF2 | 4.986 | 13.799 | 73 | 3.598 | 5.000 | 33 | 1.640 | 1.565 | 22 |
| IL33 | 5.102 | 17.583 | 71 | 3.036 | 2.914 | 17 | 0.927 | 2.863 | 23 |
| TNFα | 6.099 | 29.403 | 238 | 2.908 | 8.028 | 96 | −0.681 | 1.376 | 71 |
| IL-4 | 3.152 | 47.044 | 258 | 2.902 | 5.775 | 57 | 0.838 | 3.440 | 55 |
| IL-6 | 5.230 | 26.041 | 161 | 2.737 | 5.370 | 46 | n/a | ||
| IL-3 | 3.865 | 9.932 | 63 | 2.460 | 2.436 | 21 | n/a | ||
| Upstream Regulators | *Npc1−/−/App−/− Crb vs. WT Crb | *Npc1−/− Crb vs. WT Crb | *App−/− Crb vs. WT Crb | ||||||
|---|---|---|---|---|---|---|---|---|---|
| z-Score | −log(p) | #T.M | z-Score | −log(p) | #T.M | z-Score | −log(p) | #T.M | |
| IFN-γ | 9.324 | 38.497 | 262 | 5.432 | 21.225 | 84 | −0.152 | 2.481 | 69 |
| TNFα | 6.724 | 17.390 | 258 | 4.694 | 12.412 | 81 | −0.816 | 1.324 | 79 |
| IFN-α (group) | 6.567 | 14.712 | 84 | 2.981 | 11.699 | 33 | −1.845 | 0 | 12 |
| GM-CSF/CSF2 | 5.761 | 8.539 | 79 | 4.023 | 5.740 | 26 | −0.239 | 2.119 | 28 |
| IFN-β1 | 4.841 | 11.953 | 62 | 2.874 | 7.525 | 19 | 1.250 | 2.844 | 0 |
| IL-1β | 6.972 | 13.230 | 147 | 3.828 | 9.729 | 49 | n/a | ||
| IFN-α2 | 6.302 | 16.475 | 61 | 3.059 | 13.590 | 27 | n/a | ||
| IFN-β (group) | 4.956 | 7.919 | 31 | 3.595 | 10.769 | 18 | n/a | ||
| Function | *Npc1−/− Crb | *Npc1−/−App−/− Crb | *Npc1−/− Ctx | Npc1−/−App−/− Ctx |
|---|---|---|---|---|
| Microglial Activation | 9 | 29 | 15 | 29 |
| Antiviral Response | 15 | 56 | n/a | 41 |
| Antimicrobial Response | n/a | 83 | 31 | 73 |
| T-lymphocyte Activation | 18 | 87 | 37 | 79 |
| T-lymphocyte Chemotaxis | 6 | 25 | n/a | n/a |
| Activation of Antigen-Presenting Cells | n/a | 87 | 43 | 95 |
| Activation of Dendritic Cells | n/a | 27 | n/a | 25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez, K.L.; Shin, S.D.; Rajagopal, N.; White, J.B.; Currais, A.; Castell, D.-S.; Maher, P.; Soriano, S. A Potential Role for the Amyloid Precursor Protein in the Regulation of Interferon Signaling, Cholesterol Homeostasis, and Tau Phosphorylation in Niemann–Pick Disease Type C. Genes 2024, 15, 1066. https://doi.org/10.3390/genes15081066
Sanchez KL, Shin SD, Rajagopal N, White JB, Currais A, Castell D-S, Maher P, Soriano S. A Potential Role for the Amyloid Precursor Protein in the Regulation of Interferon Signaling, Cholesterol Homeostasis, and Tau Phosphorylation in Niemann–Pick Disease Type C. Genes. 2024; 15(8):1066. https://doi.org/10.3390/genes15081066
Chicago/Turabian StyleSanchez, Kayla L., Samuel D. Shin, Naren Rajagopal, Jacob B. White, Antonio Currais, David-Soriano Castell, Pamela Maher, and Salvador Soriano. 2024. "A Potential Role for the Amyloid Precursor Protein in the Regulation of Interferon Signaling, Cholesterol Homeostasis, and Tau Phosphorylation in Niemann–Pick Disease Type C" Genes 15, no. 8: 1066. https://doi.org/10.3390/genes15081066