
Advances in the Application of Protein Language Modeling for Nucleic Acid Protein Binding Site Prediction


Abstract
1. Introduction
2. Biological Language Model
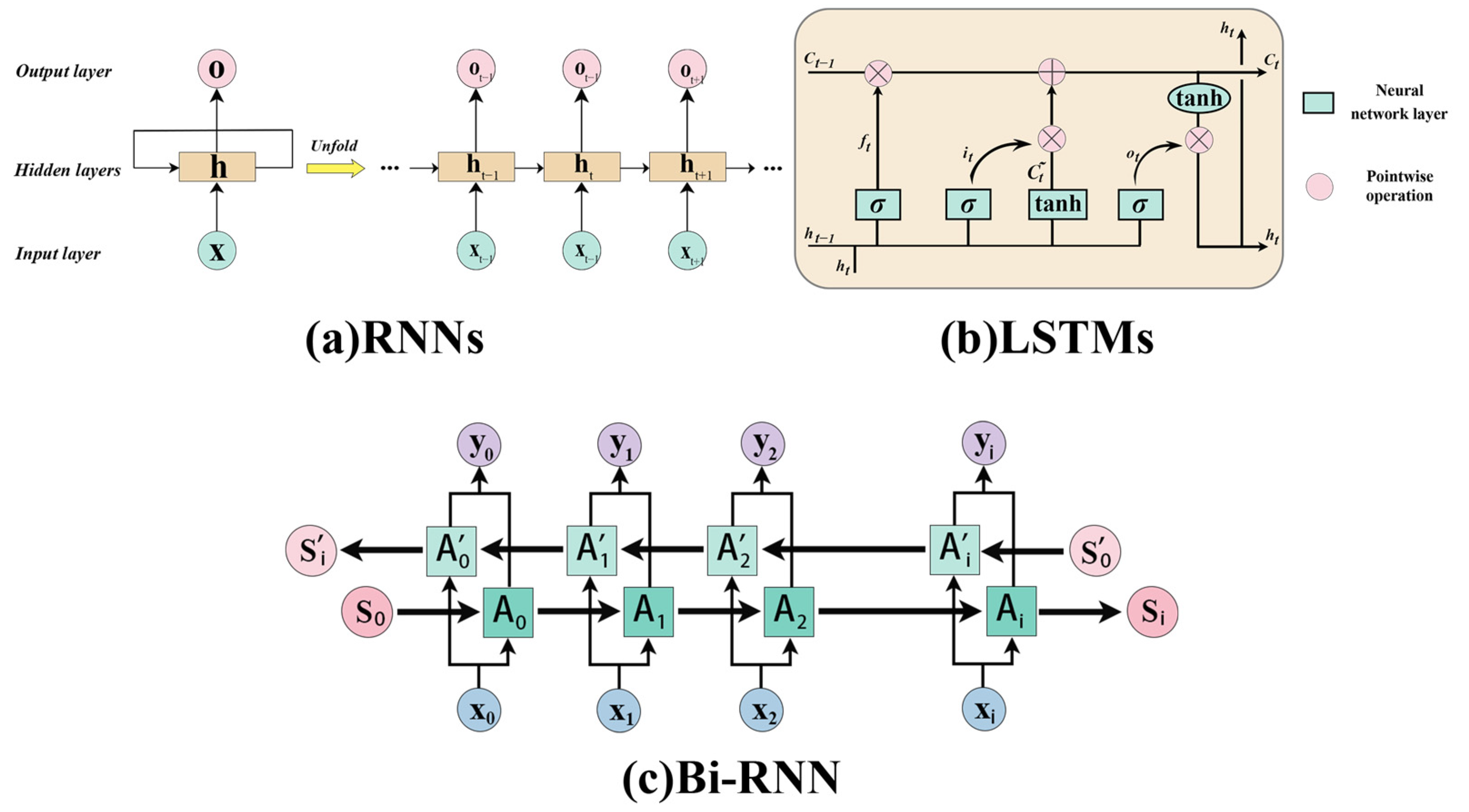
2.1. RNNs and LSTM
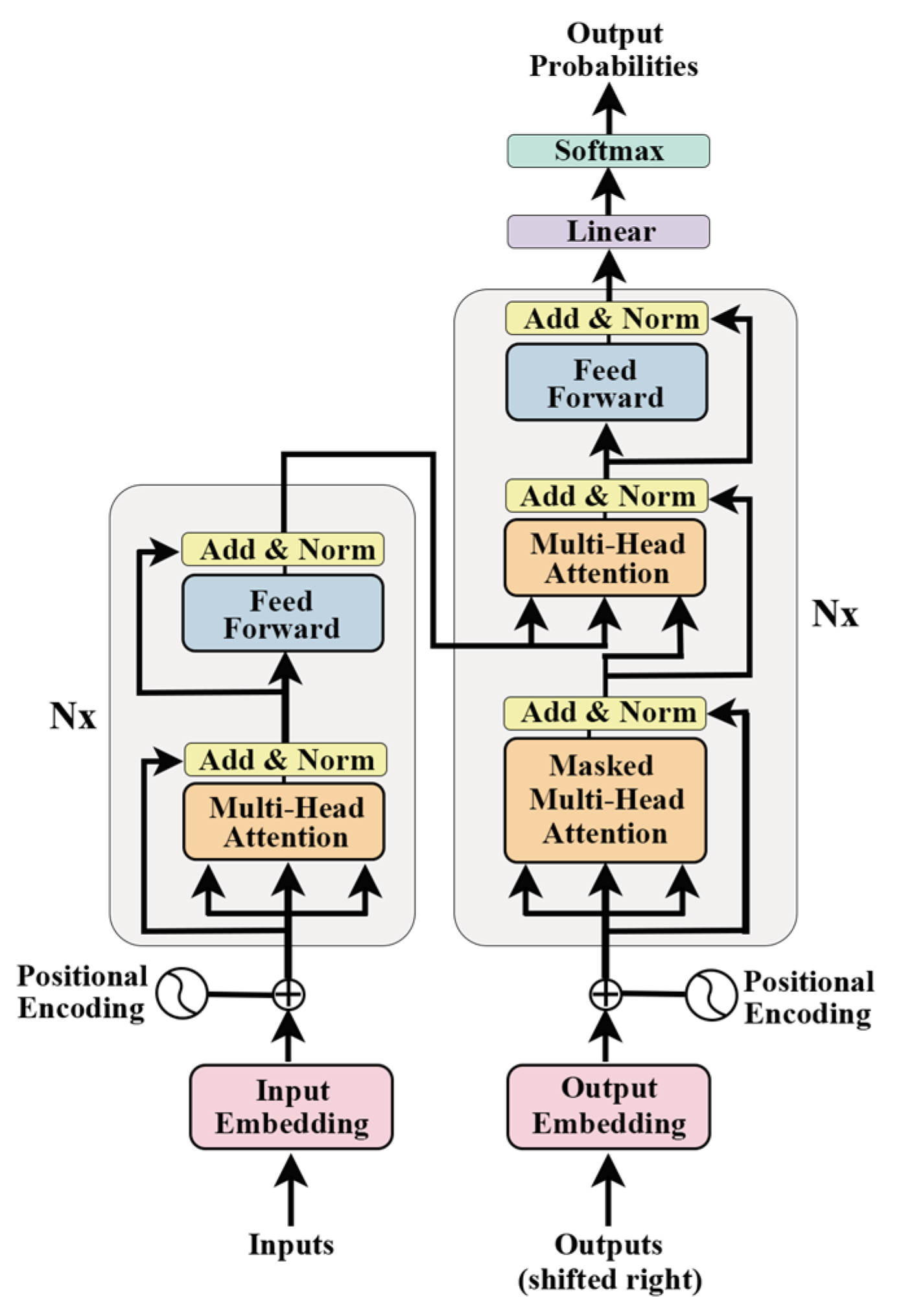
2.2. Attention Mechanism and Transformer
2.3. Protein Language Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hyperparameter | ESM-1b | ESM-MSA-1b | ESM-1v | ESM-2 |
|---|---|---|---|---|
| Dataset | UniRef50 | UniRef50 | MSA | UniRef90 |
| Number of layers | 33 | 12 | 33 | 48 |
| Params | 650 M | 100 M | 650 M | 15 B |
| Embedding Dim | 1028 | 768 | 1028 | 5120 |
| Input | Single-sequence | MSA | Single-sequence | Single-sequence |
| Universality | Family-specific | Few-shot | Zero-short | Zero-short |
| Model | Transformer | Two rows of attention mechanisms have been added | Transformer | Transformer |
| References | [69] | [49] | [74] | [75] |
| Hyperparameter | ProtTXL | ProtBert | ProtXLNet | ProtALbert | ProtElectra | ProtT5-XL | ProtT5-XXL | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Dataset | BFD 100 | BFD 100 | UniRef 100 | UniRef 100 | UniRef 100 | UniRef 100 | UniRef 100 | UniRef 50 | BFD 100 | UniRef 50 | BFD 100 |
| Number of layers | 32 | 30 | 30 | 30 | 12 | 30 | 24 | 24 | |||
| Params | 562 M | 420 M | 409 M | 409 M | 224 M | 420 M | 3 B | 11 B | |||
| Hidden layers size | 1024 | 1024 | 1028 | 1024 | 1024 | 1024 | 1024 | ||||
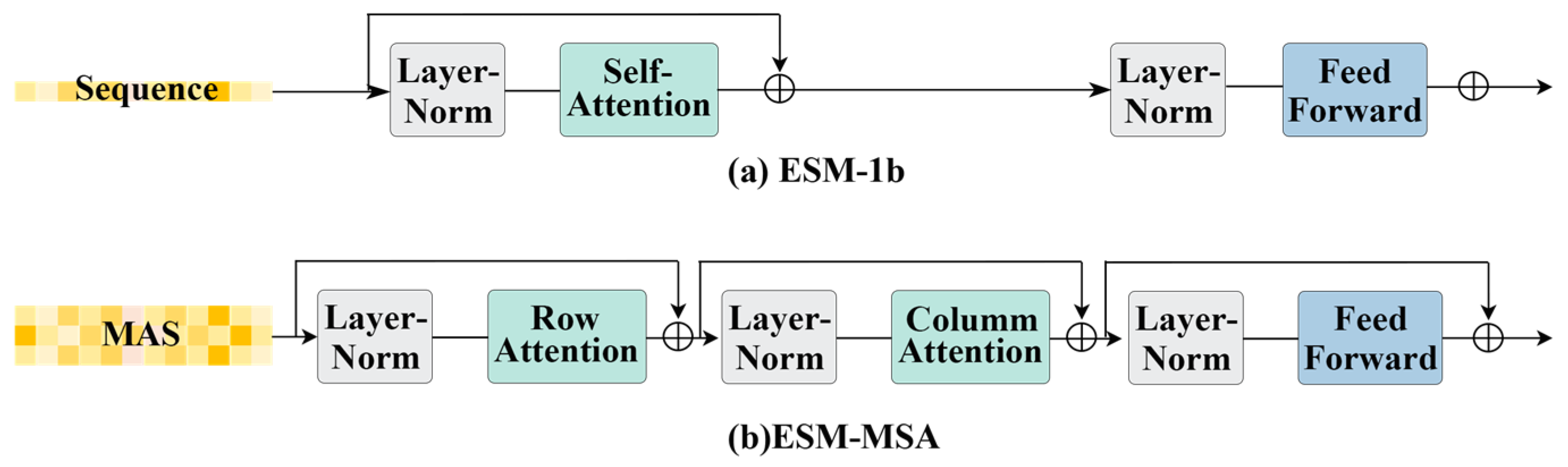
2.4. Nucleic Acid Language Models
3. Methods of Nucleic Acid Protein Binding Sites Prediction
3.1. Overview of Methods Framework
3.2. Benchmark Datasets
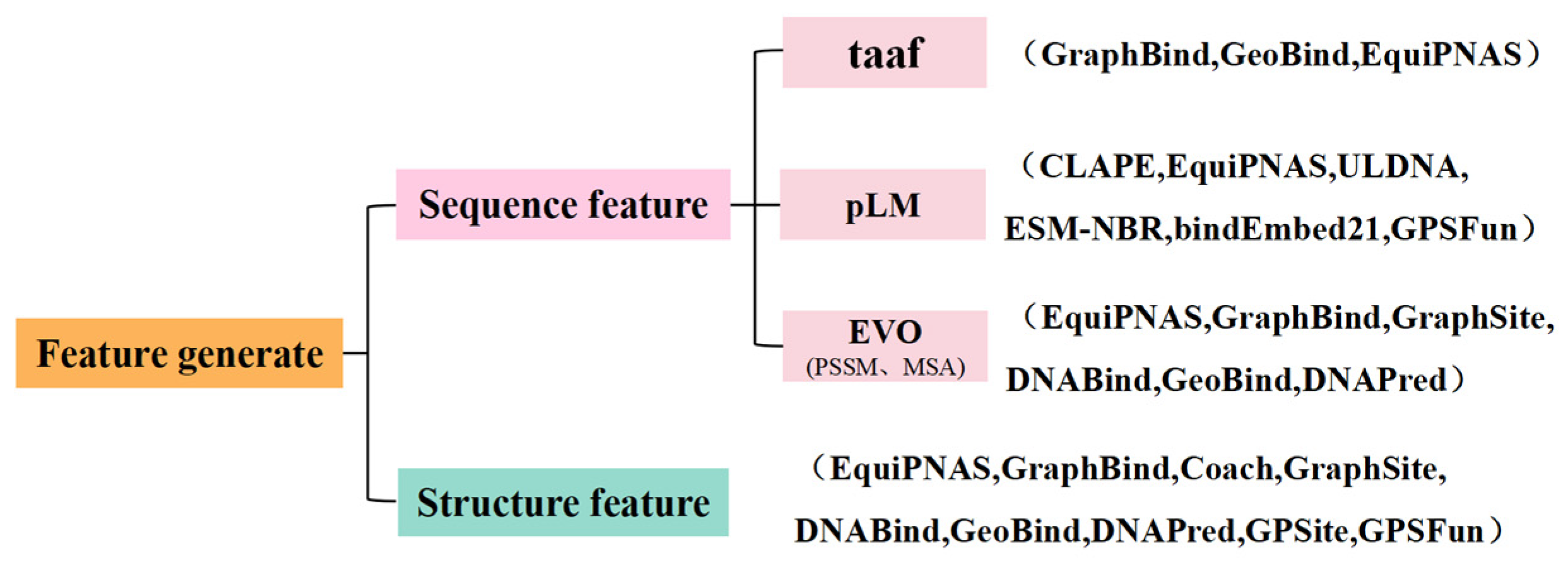
3.3. Feature Extraction
3.3.1. Features Based on Amino Acids
3.3.2. Features Based on Evolutionary Information
3.3.3. Feature Based on Structure
3.3.4. Feature Representation Extraction from pLMs
3.4. Performance Evaluation
3.5. Ablation Studies
4. Discussion
5. Conclusions
Funding
Conflicts of Interest
References
- Charoensawan, V.; Wilson, D.; Teichmann, S.A. Genomic Repertoires of DNA-Binding Transcription Factors across the Tree of Life. Nucleic Acids Res. 2010, 38, 7364–7377. [Google Scholar] [CrossRef]
- Stormo, G.D.; Zhao, Y. Determining the Specificity of Protein–DNA Interactions. Nat. Rev. Genet. 2010, 11, 751–760. [Google Scholar] [CrossRef]
- Zhang, Q.C.; Petrey, D.; Deng, L.; Qiang, L.; Shi, Y.; Thu, C.A.; Bisikirska, B.; Lefebvre, C.; Accili, D.; Hunter, T.; et al. Structure-Based Prediction of Protein–Protein Interactions on a Genome-Wide Scale. Nature 2012, 490, 556–560. [Google Scholar] [CrossRef]
- Yu, B.; Pettitt, B.M.; Iwahara, J. Dynamics of Ionic Interactions at Protein–Nucleic Acid Interfaces. Acc. Chem. Res. 2020, 53, 1802–1810. [Google Scholar] [CrossRef]
- Schmidtke, P.; Barril, X. Understanding and Predicting Druggability. A High-Throughput Method for Detection of Drug Binding Sites. J. Med. Chem. 2010, 53, 5858–5867. [Google Scholar] [CrossRef]
- Yu, Y.; Li, S.; Ser, Z.; Kuang, H.; Than, T.; Guan, D.; Zhao, X.; Patel, D.J. Cryo-EM Structure of DNA-Bound Smc5/6 Reveals DNA Clamping Enabled by Multi-Subunit Conformational Changes. Proc. Natl. Acad. Sci. USA 2022, 119, e2202799119. [Google Scholar] [CrossRef]
- Dyson, H.J. Roles of Intrinsic Disorder in Protein–Nucleic Acid Interactions. Mol. BioSyst. 2012, 8, 97–104. [Google Scholar] [CrossRef]
- Järvelin, A.I.; Noerenberg, M.; Davis, I.; Castello, A. The New (Dis)Order in RNA Regulation. Cell Commun. Signal. 2016, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Xia, C.-Q.; Pan, X.; Shen, H.-B. GraphBind: Protein Structural Context Embedded Rules Learned by Hierarchical Graph Neural Networks for Recognizing Nucleic-Acid-Binding Residues. Nucleic Acids Res. 2021, 49, e51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, Q.; Liu, B. NCBRPred: Predicting Nucleic Acid Binding Residues in Proteins Based on Multilabel Learning. Brief. Bioinform. 2021, 22, bbaa397. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-H.; Hu, J.; Song, X.-N.; Yu, D.-J. DNAPred: Accurate Identification of DNA-Binding Sites from Protein Sequence by Ensembled Hyperplane-Distance-Based Support Vector Machines. J. Chem. Inf. Model. 2019, 59, 3057–3071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ghadermarzi, S.; Katuwawala, A.; Kurgan, L. DNAgenie: Accurate Prediction of DNA-Type-Specific Binding Residues in Protein Sequences. Brief. Bioinform. 2021, 22, bbab336. [Google Scholar] [CrossRef]
- Walia, R.R.; Xue, L.C.; Wilkins, K.; El-Manzalawy, Y.; Dobbs, D.; Honavar, V. RNABindRPlus: A Predictor That Combines Machine Learning and Sequence Homology-Based Methods to Improve the Reliability of Predicted RNA-Binding Residues in Proteins. PLoS ONE 2014, 9, e97725. [Google Scholar] [CrossRef]
- Qiu, J.; Bernhofer, M.; Heinzinger, M.; Kemper, S.; Norambuena, T.; Melo, F.; Rost, B. ProNA2020 Predicts Protein–DNA, Protein–RNA, and Protein–Protein Binding Proteins and Residues from Sequence. J. Mol. Biol. 2020, 432, 2428–2443. [Google Scholar] [CrossRef]
- Armon, A.; Graur, D.; Ben-Tal, N. ConSurf: An Algorithmic Tool for the Identification of Functional Regions in Proteins by Surface Mapping of Phylogenetic Information. J. Mol. Biol. 2001, 307, 447–463. [Google Scholar] [CrossRef]
- Hu, J.; Li, Y.; Zhang, M.; Yang, X.; Shen, H.-B.; Yu, D.-J. Predicting Protein-DNA Binding Residues by Weightedly Combining Sequence-Based Features and Boosting Multiple SVMs. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 1389–1398. [Google Scholar] [CrossRef]
- Zhang, J.; Kurgan, L. SCRIBER: Accurate and Partner Type-Specific Prediction of Protein-Binding Residues from Proteins Sequences. Bioinformatics 2019, 35, i343–i353. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-J.; Hu, J.; Yang, J.; Shen, H.-B.; Tang, J.; Yang, J.-Y. Designing Template-Free Predictor for Targeting Protein-Ligand Binding Sites with Classifier Ensemble and Spatial Clustering. IEEE/ACM Trans. Comput. Biol. Bioinform. 2013, 10, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xie, Z.-R.; Wu, Y. Understand Protein Functions by Comparing the Similarity of Local Structural Environments. Biochim. Biophys. Acta 2017, 1865, 142–152. [Google Scholar] [CrossRef]
- Wu, Q.; Peng, Z.; Zhang, Y.; Yang, J. COACH-D: Improved Protein–Ligand Binding Sites Prediction with Refined Ligand-Binding Poses through Molecular Docking. Nucleic Acids Res. 2018, 46, W438–W442. [Google Scholar] [CrossRef]
- Su, H.; Liu, M.; Sun, S.; Peng, Z.; Yang, J. Improving the Prediction of Protein–Nucleic Acids Binding Residues via Multiple Sequence Profiles and the Consensus of Complementary Methods. Bioinformatics 2019, 35, 930–936. [Google Scholar] [CrossRef]
- Liu, R.; Hu, J. DNABind: A Hybrid Algorithm for Structure-Based Prediction of DNA-Binding Residues by Combining Machine Learning- and Template-Based Approaches: DNA-Binding Residue Prediction. Proteins 2013, 81, 1885–1899. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, J.; Doerr, S.; Martínez-Rosell, G.; Rose, A.S.; De Fabritiis, G. DeepSite: Protein-Binding Site Predictor Using 3D-Convolutional Neural Networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yamashita, K.; Amada, K.M.; Standley, D.M. Quantifying Sequence and Structural Features of Protein–RNA Interactions. Nucleic Acids Res. 2014, 42, 10086–10098. [Google Scholar] [CrossRef]
- Lam, J.H.; Li, Y.; Zhu, L.; Umarov, R.; Jiang, H.; Héliou, A.; Sheong, F.K.; Liu, T.; Long, Y.; Li, Y.; et al. A Deep Learning Framework to Predict Binding Preference of RNA Constituents on Protein Surface. Nat. Commun. 2019, 10, 4941. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Chen, S.; Rao, J.; Zheng, S.; Zhao, H.; Yang, Y. AlphaFold2-Aware Protein-DNA Binding Site Prediction Using Graph Transformer. Brief. Bioinform. 2022, 23, bbab564. [Google Scholar] [CrossRef]
- Roche, R.; Moussad, B.; Shuvo, M.H.; Tarafder, S.; Bhattacharya, D. EquiPNAS: Improved Protein–Nucleic Acid Binding Site Prediction Using Protein-Language-Model-Informed Equivariant Deep Graph Neural Networks. Nucleic Acids Res. 2024, 52, e27. [Google Scholar] [CrossRef]
- Abola, E.E.; Bernstein, F.C.; Koetzle, T.F. The Protein Data Bank. In Neutrons in Biology; Schoenborn, B.P., Ed.; Springer: Boston, MA, USA, 1984; p. 441. ISBN 978-1-4899-0377-8. [Google Scholar]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Applying and Improving AlphaFold at CASP14. Proteins 2021, 89, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Wang, X.; Yu, S.; Lou, E.; Tan, Y.-L.; Tan, Z.-J. RNA 3D Structure Prediction: Progress and Perspective. Molecules 2023, 28, 5532. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chiu, T.-P.; Rohs, R. Predicting DNA Structure Using a Deep Learning Method. Nat. Commun. 2024, 15, 1243. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Zhang, Y.; Xiong, Y.; Xiao, Y. Advances in RNA 3D Structure Prediction. J. Chem. Inf. Model. 2022, 62, 5862–5874. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Sweeney, B.A.; Bateman, A.; Cerny, J.; Zok, T.; Szachniuk, M. When Will RNA Get Its AlphaFold Moment? Nucleic Acids Res. 2023, 51, 9522–9532. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Antczak, M.; Szachniuk, M.; Zok, T.; Kretsch, R.C.; Rangan, R.; Pham, P.; Das, R.; Robin, X.; Studer, G.; et al. New Prediction Categories in CASP15. Proteins 2023, 91, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hu, Z.; Sun, S.; Tan, Q.; Wang, Y.; Yu, Q.; Zong, L.; Hong, L.; Xiao, J.; Shen, T.; et al. Interpretable RNA Foundation Model from Unannotated Data for Highly Accurate RNA Structure and Function Predictions. arXiv 2022, arXiv:2204.00300. [Google Scholar]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-Scale Prediction of Atomic-Level Protein Structure with a Language Model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Littmann, M.; Heinzinger, M.; Dallago, C.; Weissenow, K.; Rost, B. Protein Embeddings and Deep Learning Predict Binding Residues for Various Ligand Classes. Sci. Rep. 2021, 11, 23916. [Google Scholar] [CrossRef]
- Zhu, Y.-H.; Zhang, C.; Yu, D.-J.; Zhang, Y. Integrating Unsupervised Language Model with Triplet Neural Networks for Protein Gene Ontology Prediction. PLoS Comput. Biol. 2022, 18, e1010793. [Google Scholar] [CrossRef]
- Madani, A.; Krause, B.; Greene, E.R.; Subramanian, S.; Mohr, B.P.; Holton, J.M.; Olmos, J.L.; Xiong, C.; Sun, Z.Z.; Socher, R.; et al. Large Language Models Generate Functional Protein Sequences across Diverse Families. Nat. Biotechnol. 2023, 41, 1099–1106. [Google Scholar] [CrossRef]
- Ferruz, N.; Schmidt, S.; Höcker, B. ProtGPT2 Is a Deep Unsupervised Language Model for Protein Design. Nat. Commun. 2022, 13, 4348. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yuan, Q.; Zhao, H.; Yang, Y. Accurately Identifying Nucleic-Acid-Binding Sites through Geometric Graph Learning on Language Model Predicted Structures. Brief. Bioinform. 2023, 24, bbad360. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Shen, Y.-Y.; Liu, R. Structure-Based Prediction of Nucleic Acid Binding Residues by Merging Deep Learning- and Template-Based Approaches. PLoS Comput. Biol. 2023, 19, e1011428. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; McHugh, R.; Anishchenko, I.; Jiang, H.; Baker, D.; DiMaio, F. Accurate Prediction of Protein–Nucleic Acid Complexes Using RoseTTAFoldNA. Nat. Methods 2024, 21, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Bhattacharya, N.; Thomas, N.; Duan, Y.; Chen, X.; Canny, J.; Abbeel, P.; Song, Y.S. Evaluating Protein Transfer Learning with TAPE. In Proceedings of the Advances in Neural Information Processing Systems 32 (NeurIPS 2019), Vancouver, BC, Canada, 8–14 December 2019. [Google Scholar]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning. IEEE Trans. Pattern Anal. Mach. Intell. 2021, 44, 7112–7127. [Google Scholar] [CrossRef] [PubMed]
- Heinzinger, M.; Elnaggar, A.; Wang, Y.; Dallago, C.; Nechaev, D.; Matthes, F.; Rost, B. Modeling Aspects of the Language of Life through Transfer-Learning Protein Sequences. BMC Bioinform. 2019, 20, 723. [Google Scholar] [CrossRef]
- Rao, R.; Liu, J.; Verkuil, R.; Meier, J.; Canny, J.F.; Abbeel, P.; Sercu, T.; Rives, A. MSA Transformer. In Proceedings of the International Conference on Machine Learning, Virtual, 18–24 July 2021. [Google Scholar]
- Fang, Y.; Jiang, Y.; Wei, L.; Ma, Q.; Ren, Z.; Yuan, Q.; Wei, D.-Q. DeepProSite: Structure-Aware Protein Binding Site Prediction Using ESMFold and Pretrained Language Model. Bioinformatics 2023, 39, btad718. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-H.; Liu, Z.; Liu, Y.; Ji, Z.; Yu, D.-J. ULDNA: Integrating Unsupervised Multi-Source Language Models with LSTM-Attention Network for High-Accuracy Protein–DNA Binding Site Prediction. Brief. Bioinform. 2024, 25, bbae040. [Google Scholar] [CrossRef]
- Zeng, W.; Lv, D.; Liu, X.; Chen, G.; Liu, W.; Peng, S. ESM-NBR: Fast and Accurate Nucleic Acid-Binding Residue Prediction via Protein Language Model Feature Representation and Multi-Task Learning. In Proceedings of the 2023 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Istanbul, Turkiye, 5–8 December 2023; pp. 76–81. [Google Scholar]
- Liu, Y.; Tian, B. Protein–DNA Binding Sites Prediction Based on Pre-Trained Protein Language Model and Contrastive Learning. Brief. Bioinform. 2023, 25, bbad488. [Google Scholar] [CrossRef] [PubMed]
- Bepler, T.; Berger, B. Learning the Protein Language: Evolution, Structure, and Function. Cell Syst. 2021, 12, 654–669.e3. [Google Scholar] [CrossRef]
- Lample, G.; Ballesteros, M.; Subramanian, S.; Kawakami, K.; Dyer, C. Neural Architectures for Named Entity Recognition. arXiv 2016, arXiv:1603.01360. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention Is All You Need. arXiv 2017, arXiv:1706.03762. [Google Scholar]
- Shen, Y.; Chen, Z.; Mamalakis, M.; He, L.; Xia, H.; Li, T.; Su, Y.; He, J.; Wang, Y.G. A Fine-Tuning Dataset and Benchmark for Large Language Models for Protein Understanding. arXiv 2024, arXiv:2406.05540. [Google Scholar]
- Graves, A.; Liwicki, M.; Fernandez, S.; Bertolami, R.; Bunke, H.; Schmidhuber, J. A Novel Connectionist System for Unconstrained Handwriting Recognition. IEEE Trans. Pattern Anal. Mach. Intell. 2009, 31, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Xia, J.; Zheng, J.; Tan, C.; Huang, Y.; Xu, Y.; Li, S.Z. Protein Language Models and Structure Prediction: Connection and Progression. arXiv 2022, arXiv:2211.16742. [Google Scholar]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. BERT: Pre-Training of Deep Bidirectional Transformers for Language Understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Radford, A.; Narasimhan, K.; Salimans, T.; Sutskever, I. Improving Language Understanding by Generative Pre-Training. Available online: https://cdn.openai.com/research-covers/language-unsupervised/language_understanding_paper.pdf (accessed on 22 July 2018).
- Brown, T.B.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.; Dhariwal, P.; Neelakantan, A.; Shyam, P.; Sastry, G.; Askell, A.; et al. Language Models Are Few-Shot Learners. In Proceedings of the Advances in Neural Information Processing Systems 33 (NeurIPS 2020), Virtual, 6–12 December 2020. [Google Scholar]
- Wang, S.; Peng, J.; Ma, J.; Xu, J. Protein Secondary Structure Prediction Using Deep Convolutional Neural Fields. Sci. Rep. 2016, 6, 18962. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, R.; Paliwal, K.; Lyons, J.; Singh, J.; Yang, Y.; Zhou, Y. Single-sequence-based Prediction of Protein Secondary Structures and Solvent Accessibility by Deep Whole-sequence Learning. J. Comput. Chem. 2018, 39, 2210–2216. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, Y. OCLSTM: Optimized Convolutional and Long Short-Term Memory Neural Network Model for Protein Secondary Structure Prediction. PLoS ONE 2021, 16, e0245982. [Google Scholar] [CrossRef]
- Heffernan, R.; Yang, Y.; Paliwal, K.; Zhou, Y. Capturing Non-Local Interactions by Long Short-Term Memory Bidirectional Recurrent Neural Networks for Improving Prediction of Protein Secondary Structure, Backbone Angles, Contact Numbers and Solvent Accessibility. Bioinformatics 2017, 33, 2842–2849. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Zou, K.; Zhang, Z.; Yang, F. GLTM: A Global-Local Attention LSTM Model to Locate Dimer Motif of Single-Pass Membrane Proteins. Front. Genet. 2022, 13, 854571. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Shen, Q.; Zhang, G.; Wang, P.; Yu, Z.-G. LSTMCNNsucc: A Bidirectional LSTM and CNN-Based Deep Learning Method for Predicting Lysine Succinylation Sites. BioMed Res. Int. 2021, 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological Structure and Function Emerge from Scaling Unsupervised Learning to 250 Million Protein Sequences. Proc. Natl. Acad. Sci. USA 2021, 118, e2016239118. [Google Scholar] [CrossRef] [PubMed]
- Strodthoff, N.; Wagner, P.; Wenzel, M.; Samek, W. UDSMProt: Universal Deep Sequence Models for Protein Classification. Bioinformatics 2020, 36, 2401–2409. [Google Scholar] [CrossRef]
- Alley, E.C.; Khimulya, G.; Biswas, S.; AlQuraishi, M.; Church, G.M. Unified Rational Protein Engineering with Sequence-Based Deep Representation Learning. Nat. Methods 2019, 16, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Chatzou, M.; Magis, C.; Chang, J.-M.; Kemena, C.; Bussotti, G.; Erb, I.; Notredame, C. Multiple Sequence Alignment Modeling: Methods and Applications. Brief. Bioinform. 2016, 17, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. ProteinBERT: A Universal Deep-Learning Model of Protein Sequence and Function. Bioinformatics 2022, 38, 2102–2110. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.; Rao, R.; Verkuil, R.; Liu, J.; Sercu, T.; Rives, A. Language Models Enable Zero-Shot Prediction of the Effects of Mutations on Protein Function. In Proceedings of the Advances in Neural Information Processing Systems 34 (NeurIPS 2021), Virtual, 6–14 December 2021. [Google Scholar]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Fazel-Zarandi, M.; Sercu, T.; Candido, S.; Rives, A. Language Models of Protein Sequences at the Scale of Evolution Enable Accurate Structure Prediction. BioRxiv 2022, 2022, 500902. [Google Scholar] [CrossRef]
- Ji, Y.; Zhou, Z.; Liu, H.; Davuluri, R.V. DNABERT: Pre-Trained Bidirectional Encoder Representations from Transformers Model for DNA-Language in Genome. Bioinformatics 2021, 37, 2112–2120. [Google Scholar] [CrossRef]
- Bernard, C.; Postic, G.; Ghannay, S.; Tahi, F. RNA-TorsionBERT: Leveraging Language Models for RNA 3D Torsion Angles Prediction. bioRxiv 2024, 597803. [Google Scholar] [CrossRef]
- Zhang, Z.; Sabuncu, M. Generalized Cross Entropy Loss for Training Deep Neural Networks with Noisy Labels. In Proceedings of the Advances in Neural Information Processing Systems 31 (NeurIPS 2018), Montréal, QC, Canada, 3–8 December 2018. [Google Scholar]
- He, X.; Zhou, Y.; Zhou, Z.; Bai, S.; Bai, X. Triplet-Center Loss for Multi-View 3D Object Retrieval. In Proceedings of the 2018 IEEE/CVF Conference on Computer Vision and Pattern Recognition, Salt Lake City, UT, USA, 18–23 June 2018; pp. 1945–1954. [Google Scholar]
- Yang, J.; Roy, A.; Zhang, Y. BioLiP: A Semi-Manually Curated Database for Biologically Relevant Ligand–Protein Interactions. Nucleic Acids Res. 2012, 41, D1096–D1103. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, S.; Madden, T.L. BLAST: At the Core of a Powerful and Diverse Set of Sequence Analysis Tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. TM-Align: A Protein Structure Alignment Algorithm Based on the TM-Score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Gromiha, M.M.; Sarai, A. Real Value Prediction of Solvent Accessibility from Amino Acid Sequence. Proteins 2003, 50, 629–635. [Google Scholar] [CrossRef]
- Pande, A.; Patiyal, S.; Lathwal, A.; Arora, C.; Kaur, D.; Dhall, A.; Mishra, G.; Kaur, H.; Sharma, N.; Jain, S.; et al. Computing Wide Range of Protein/Peptide Features from Their Sequence and Structure. BioRxiv 2019, 599126. [Google Scholar] [CrossRef]
- Patiyal, S.; Dhall, A.; Raghava, G.P.S. A Deep Learning-Based Method for the Prediction of DNA Interacting Residues in a Protein. Brief. Bioinform. 2022, 23, bbac322. [Google Scholar] [CrossRef]
- Li, P.; Liu, Z.-P. GeoBind: Segmentation of Nucleic Acid Binding Interface on Protein Surface with Geometric Deep Learning. Nucleic Acids Res. 2023, 51, e60. [Google Scholar] [CrossRef]
- Schaffer, A.A. Improving the Accuracy of PSI-BLAST Protein Database Searches with Composition-Based Statistics and Other Refinements. Nucleic Acids Res. 2001, 29, 2994–3005. [Google Scholar] [CrossRef]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-Fast Iterative Protein Sequence Searching by HMM-HMM Alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Katoh, K. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, M.; Söding, J. MMseqs2 Enables Sensitive Protein Sequence Searching for the Analysis of Massive Data Sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Richards, F.M. The Interpretation of Protein Structures: Estimation of Static Accessibility. J. Mol. Biol. 1971, 55, 379-IN4. [Google Scholar] [CrossRef]
- Joo, K.; Lee, S.J.; Lee, J. Sann: Solvent Accessibility Prediction of Proteins by Nearest Neighbor Method. Proteins 2012, 80, 1791–1797. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Faraggi, E.; Zhang, T.; Yang, Y.; Kurgan, L.; Zhou, Y. SPINE X: Improving Protein Secondary Structure Prediction by Multistep Learning Coupled with Prediction of Solvent Accessible Surface Area and Backbone Torsion Angles. J. Comput. Chem. 2012, 33, 259–267. [Google Scholar] [CrossRef]
- Yuan, Q.; Tian, C.; Yang, Y. Genome-Scale Annotation of Protein Binding Sites via Language Model and Geometric Deep Learning. eLife 2024, 13, RP93695. [Google Scholar] [CrossRef]
- Yuan, Q.; Tian, C.; Song, Y.; Ou, P.; Zhu, M.; Zhao, H.; Yang, Y. GPSFun: Geometry-Aware Protein Sequence Function Predictions with Language Models. Nucleic Acids Res. 2024, 52, W248–W255. [Google Scholar] [CrossRef] [PubMed]
- Suzek, B.E.; Huang, H.; McGarvey, P.; Mazumder, R.; Wu, C.H. UniRef: Comprehensive and Non-Redundant UniProt Reference Clusters. Bioinformatics 2007, 23, 1282–1288. [Google Scholar] [CrossRef]
- Steinegger, M.; Mirdita, M.; Söding, J. Protein-Level Assembly Increases Protein Sequence Recovery from Metagenomic Samples Manyfold. Nat. Methods 2019, 16, 603–606. [Google Scholar] [CrossRef]
- Wolf, T.; Debut, L.; Sanh, V.; Chaumond, J.; Delangue, C.; Moi, A.; Cistac, P.; Rault, T.; Louf, R.; Funtowicz, M.; et al. HuggingFace’s Transformers: State-of-the-Art Natural Language Processing. arXiv 2019, arXiv:1910.03771. [Google Scholar]
- Yan, J.; Kurgan, L. DRNApred, Fast Sequence-Based Method That Accurately Predicts and Discriminates DNA- and RNA-Binding Residues. Nucleic Acids Res. 2017, 45, e84. [Google Scholar] [CrossRef]
- Nijkamp, E.; Ruffolo, J.; Weinstein, E.N.; Naik, N.; Madani, A. ProGen2: Exploring the Boundaries of Protein Language Models. Cell Syst. 2023, 14, 968–978.e3. [Google Scholar] [CrossRef]
- Zhang, Y.; Lang, M.; Jiang, J.; Gao, Z.; Xu, F.; Litfin, T.; Chen, K.; Singh, J.; Huang, X.; Song, G.; et al. Multiple Sequence Alignment-Based RNA Language Model and Its Application to Structural Inference. Nucleic Acids Res. 2024, 52, e3. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-L.; Pang, Y.-H.; Liu, B. BioSeq-BLM: A Platform for Analyzing DNA, RNA and Protein Sequences Based on Biological Language Models. Nucleic Acids Res. 2021, 49, e129. [Google Scholar] [CrossRef]
- Zheng, M.; Sun, G.; Li, X.; Fan, Y. EGPDI: Identifying Protein–DNA Binding Sites Based on Multi-View Graph Embedding Fusion. Brief. Bioinform. 2024, 25, bbae330. [Google Scholar] [CrossRef]
- Minh, D.; Wang, H.X.; Li, Y.F.; Nguyen, T.N. Explainable Artificial Intelligence: A Comprehensive Review. Artif. Intell. Rev. 2022, 55, 3503–3568. [Google Scholar] [CrossRef]
- Jiménez-Luna, J.; Grisoni, F.; Schneider, G. Drug Discovery with Explainable Artificial Intelligence. Nat. Mach. Intell. 2020, 2, 573–584. [Google Scholar] [CrossRef]
- Nerín-Fonz, F.; Cournia, Z. Machine Learning Approaches in Predicting Allosteric Sites. Curr. Opin. Struct. Biol. 2024, 85, 102774. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Kurgan, L. High-Throughput Prediction of RNA, DNA and Protein Binding Regions Mediated by Intrinsic Disorder. Nucleic Acids Res. 2015, 43, e121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhao, B.; Shi, W.; Li, M.; Kurgan, L. DeepDISOBind: Accurate Prediction of RNA-, DNA- and Protein-Binding Intrinsically Disordered Residues with Deep Multi-Task Learning. Brief. Bioinform. 2022, 23, bbab521. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Kihara, D.; Kurgan, L. Computational Prediction of Disordered Binding Regions. Comput. Struct. Biotechnol. J. 2023, 21, 1487–1497. [Google Scholar] [CrossRef] [PubMed]
- Katuwawala, A.; Kurgan, L. Comparative Assessment of Intrinsic Disorder Predictions with a Focus on Protein and Nucleic Acid-Binding Proteins. Biomolecules 2020, 10, 1636. [Google Scholar] [CrossRef]
- Zhang, J.; Basu, S.; Kurgan, L. HybridDBRpred: Improved Sequence-Based Prediction of DNA-Binding Amino Acids Using Annotations from Structured Complexes and Disordered Proteins. Nucleic Acids Res. 2024, 52, e10. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically Disordered Proteins in Cellular Signalling and Regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]

| Method | Feature Generation | Feature Representation | Key Learning Architecture |
|---|---|---|---|
| CLAPE [53] | ProtBert | Tensor | Concatenated ACNNs |
| ESM-NBR [52] | ESM2 | Tensor | LSTM |
| DeepProSite [50] | ProtBert, DSSP | Graphs | GNN |
| EquiPNAS [27] | ESM2, DSSP, PSSM, MSA, taaf, SS, RSA, et al. | Graphs | GNN |
| ULDNA [51] | ESM, ESM-MSA, ProtBert | Tensor | LSTM |
| GPSite [99] | ProtBert, ESMFold | Graphs | GNN |
| GPSFun [100] | ProtBert, ESMFold | Graphs | GNN |
| ULDNA [51] | DeepProsite [50] | EquiPNAS [27] |
|---|---|---|
| ProtTrans + ESM-MSA ESM2 + ESM-MSA ESM2 + ProtTrans ESM2 + ProtTrans + ESM-MSA (ULDNA) | EVO DSSP ProtT5 ProtT5 + EVO + DSSP EVO + DSSP ProtT5 + EVO ProtT5 + DSSP (DeepProSite) | No ESM2 No (PSSM + MSA) No MSA No PSSM ESM2, DSSP, PSSM, MSA, taaf, SS, RSA, et al. (EquiPNAS) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Li, W. Advances in the Application of Protein Language Modeling for Nucleic Acid Protein Binding Site Prediction. Genes 2024, 15, 1090. https://doi.org/10.3390/genes15081090
Wang B, Li W. Advances in the Application of Protein Language Modeling for Nucleic Acid Protein Binding Site Prediction. Genes. 2024; 15(8):1090. https://doi.org/10.3390/genes15081090
Chicago/Turabian StyleWang, Bo, and Wenjin Li. 2024. "Advances in the Application of Protein Language Modeling for Nucleic Acid Protein Binding Site Prediction" Genes 15, no. 8: 1090. https://doi.org/10.3390/genes15081090
APA StyleWang, B., & Li, W. (2024). Advances in the Application of Protein Language Modeling for Nucleic Acid Protein Binding Site Prediction. Genes, 15(8), 1090. https://doi.org/10.3390/genes15081090