

Transcriptome Analysis Identifies Key Genes Involved in Response and Recovery to High Heat Stress Induced by Fire in Schima superba

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatments
2.2. RNA Extraction, Library Construction, and Transcriptome Sequencing
2.3. Transcriptome Assembly and Unigene Annotation
2.4. Gene Expression Quantification and Differentially Expressed Genes Analysis
2.5. Quantitative Real-Time PCR (qRT-PCR) Validation
3. Results
3.1. Statistics of the Transcriptome Sequencing in S. superba
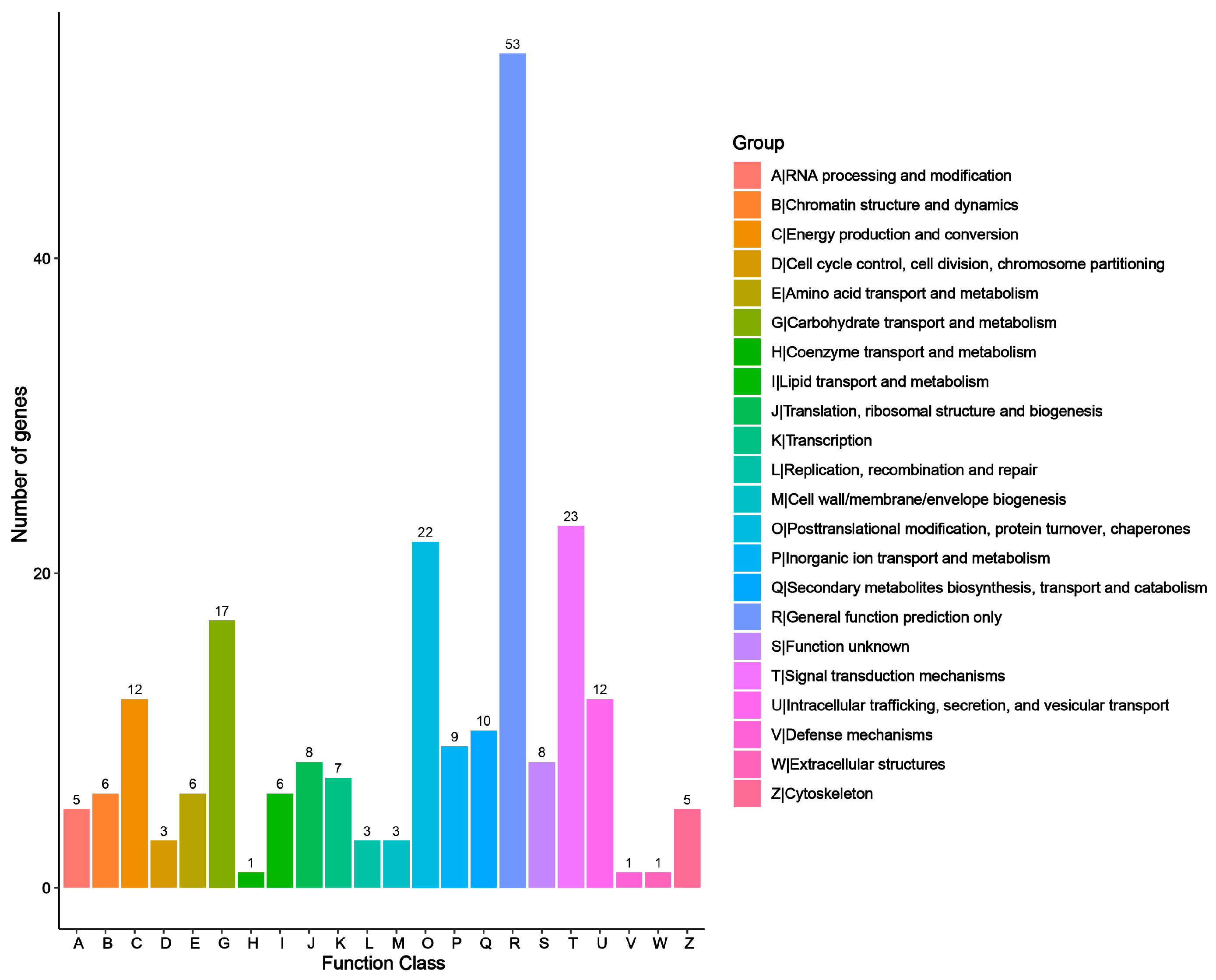
3.2. Unigene Annotation and Transcription Factors (TFs) Identification
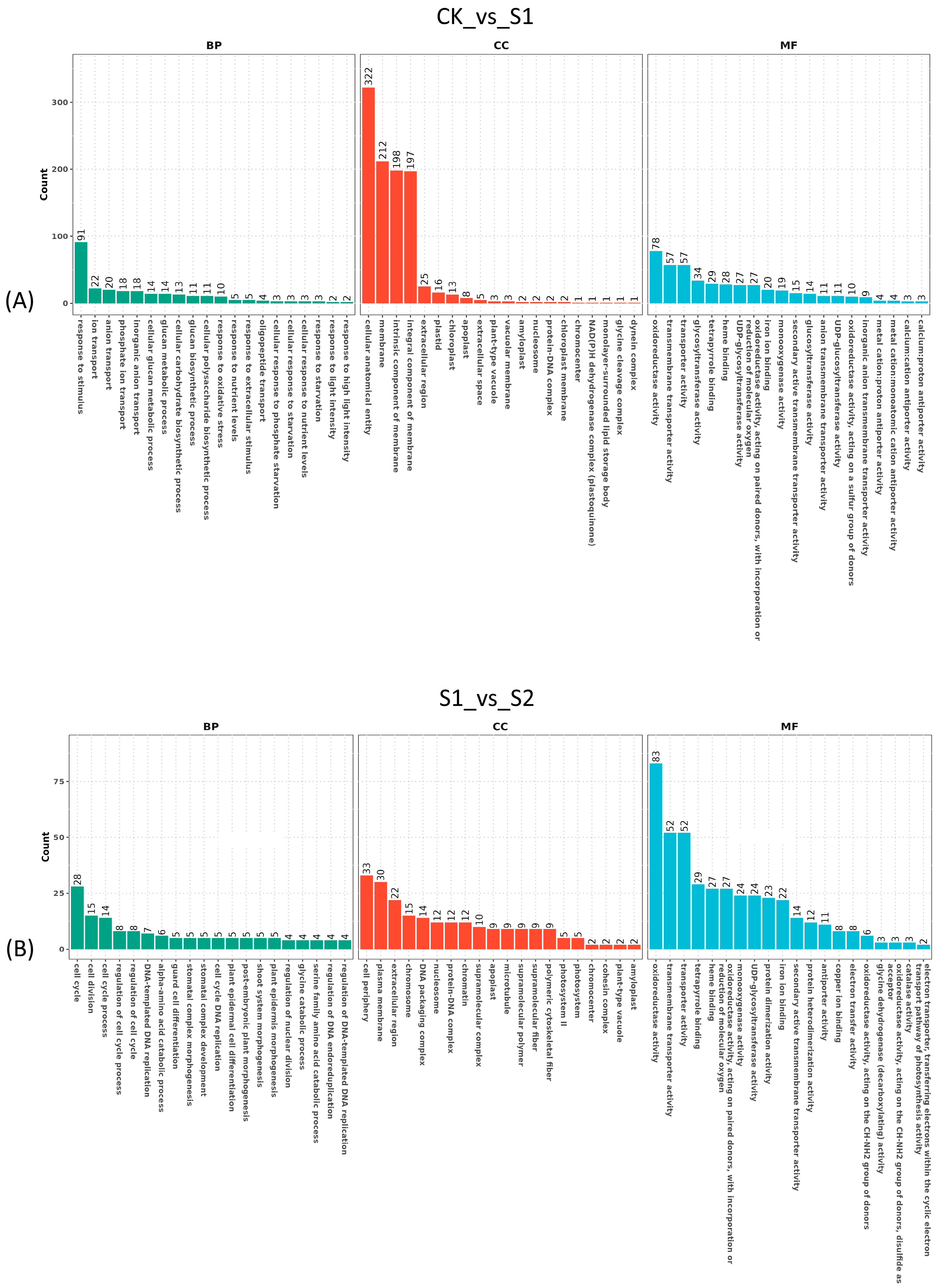
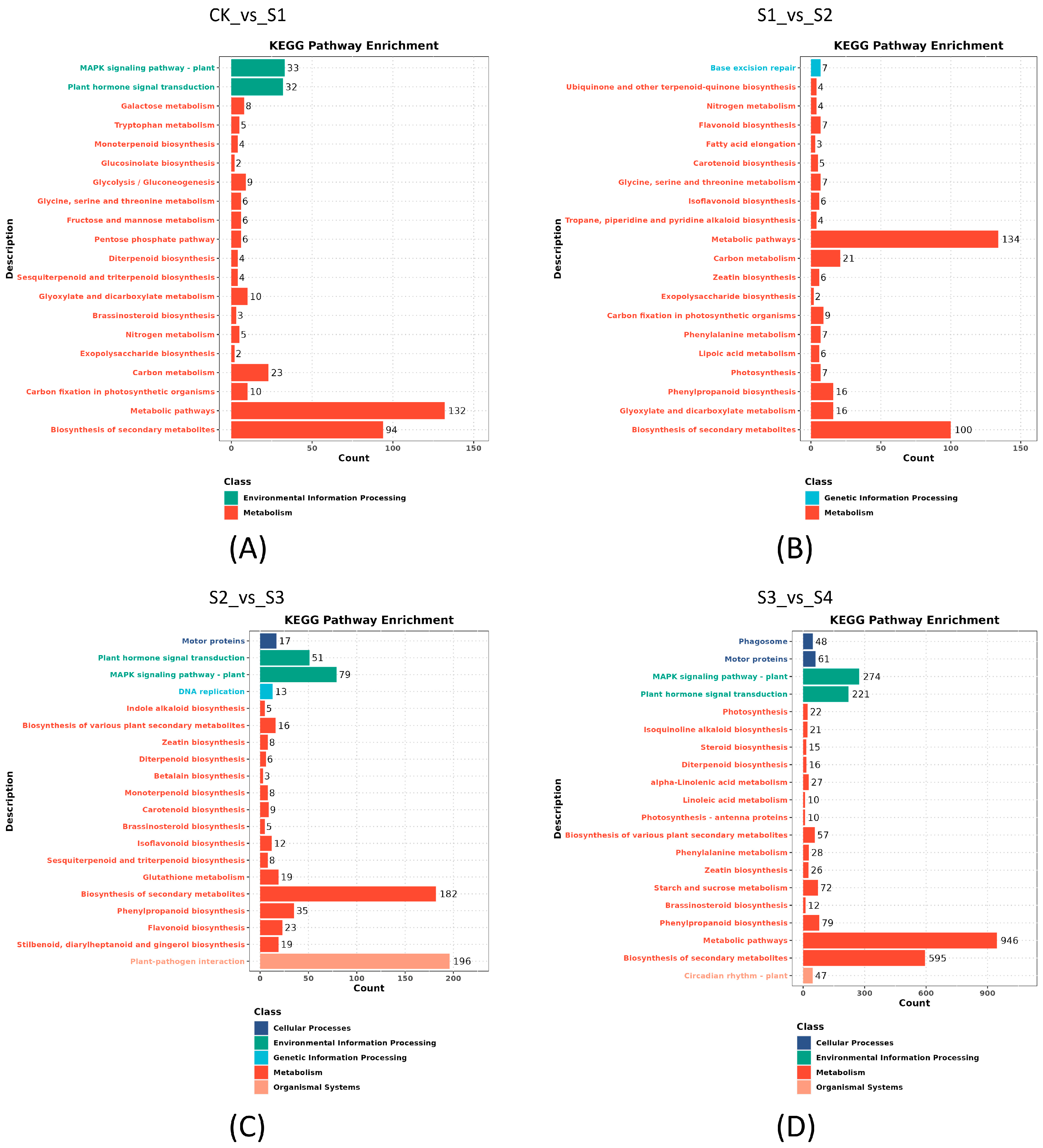
3.3. Unigenes Quantification and Differentially Expressed Genes (DEGs) Analysis
3.4. Differentially Expressed Genes Response to Strong Heat Stress in S. superba
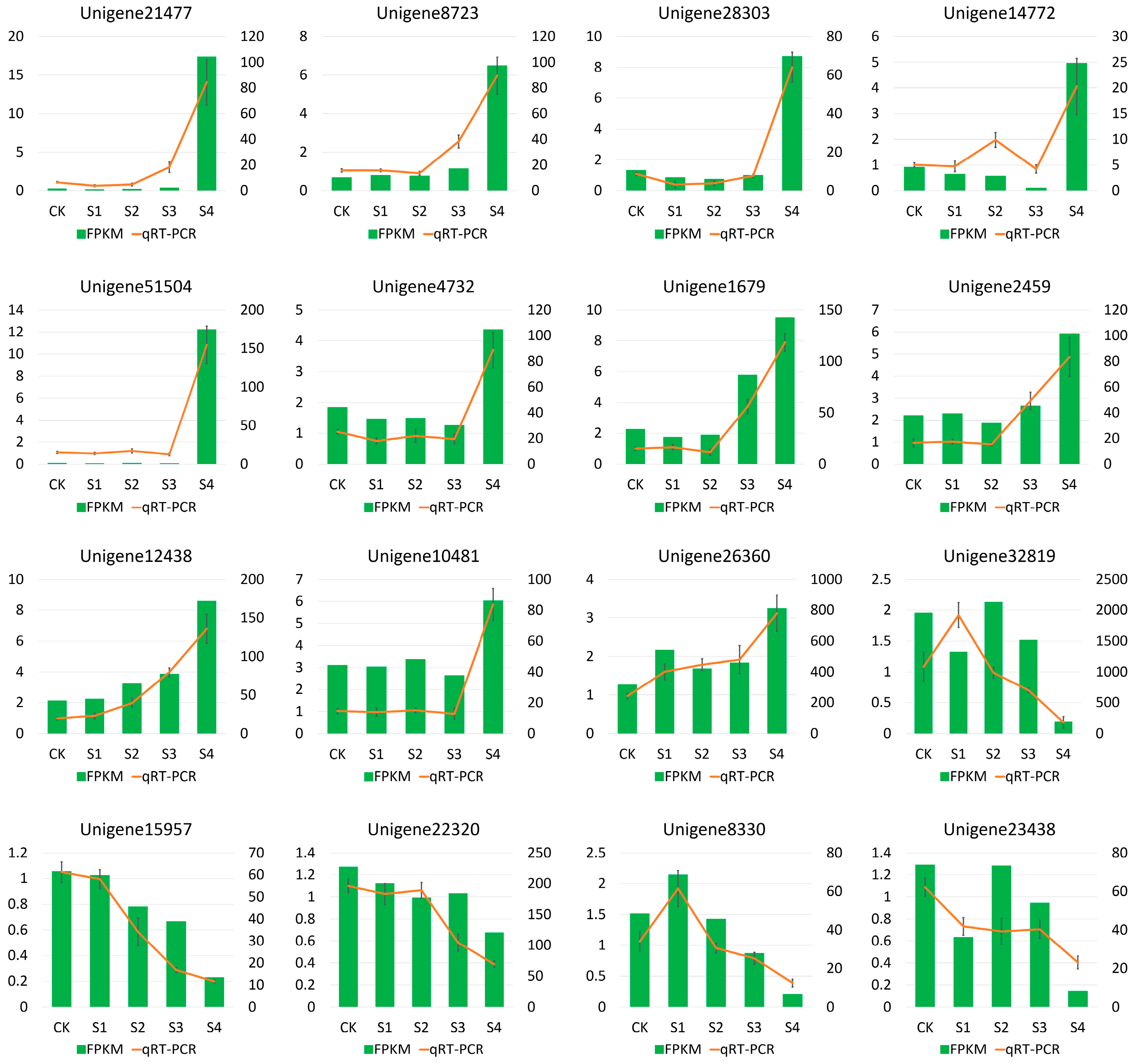
3.5. qRT-PCR Validation of DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rocca, M.E.; Brown, P.M.; MacDonald, L.H.; Carrico, C.M. Climate change impacts on fire regimes and key ecosystem services in Rocky Mountain forests. For. Ecol. Manag. 2014, 327, 290–305. [Google Scholar] [CrossRef]
- Singh, S. Forest fire emissions: A contribution to global climate change. Front. For. Glob. Chang. 2022, 5, 925480. [Google Scholar] [CrossRef]
- Noss, R.F.; Franklin, J.F.; Baker, W.L.; Schoennagel, T.; Moyle, P.B. Ecology and management of fire-prone forests of the western United States. In Society for Conservation Biology Scientific Panel or Fire in Western US Forests; Society for Conservation Biology, North American Section: Arlington, VA, USA, 2006. [Google Scholar]
- Zahed, M.; Bączek-Kwinta, R. The impact of post-fire smoke on plant communities: A global approach. Plants 2023, 12, 3835. [Google Scholar] [CrossRef] [PubMed]
- Midgley, J.J.; Bond, W.J. Plant adaptations to fire: An evolutionary perspective. In Fire Phenomena and the Earth System: An Interdisciplinary Guide to Fire Science; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 125–134. [Google Scholar]
- Odhiambo, B.O. The Effect of Fire Damage on the Growth and Survival Mechanisms of Selected Native and Commercial Trees in South Africa. Doctoral Dissertation, Stellenbosch University, Stellenbosch, South Africa, 2015. [Google Scholar]
- Rosell, J.A. Bark in woody plants: Understanding the diversity of a multifunctional structure. Integr. Comp. Biol. 2019, 59, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Kregel, K.C. Invited review: Heat shock proteins: Modifying factors in physiological stress responses and acquired thermotolerance. J. Appl. Physiol. 2002, 92, 2177–2186. [Google Scholar] [CrossRef]
- Archana, P.; Aleena, J.; Pragna, P.; Vidya, M.; Niyas, A.; Bagath, M.; Krishnan, G.; Manimaran, A.; Beena, V.; Kurien, E. Role of heat shock proteins in livestock adaptation to heat stress. J. Dairy Vet. Anim. Res. 2017, 5, 00127. [Google Scholar]
- Bokszczanin, K.L.; Consortium, S.P.T.I.T.N.; Fragkostefanakis, S. Perspectives on deciphering mechanisms underlying plant heat stress response and thermotolerance. Front. Plant Sci. 2013, 4, 315. [Google Scholar] [CrossRef] [PubMed]
- Fragkostefanakis, S.; Roeth, S.; Schleiff, E.; SCHARF, K.D. Prospects of engineering thermotolerance in crops through modulation of heat stress transcription factor and heat shock protein networks. Plant Cell Environ. 2015, 38, 1881–1895. [Google Scholar] [CrossRef]
- Liu, G.T.; Wang, J.F.; Cramer, G.; Dai, Z.W.; Duan, W.; Xu, H.G.; Wu, B.H.; Fan, P.G.; Wang, L.J.; Li, S.H. Transcriptomic analysis of grape (Vitis vinifera L.) leaves during and after recovery from heat stress. BMC Plant Biol. 2012, 12, 174. [Google Scholar] [CrossRef]
- Liang, M.H.; Jiang, J.G.; Wang, L.; Zhu, J. Transcriptomic insights into the heat stress response of Dunaliella bardawil. Enzyme Microb. Technol. 2020, 132, 109436. [Google Scholar] [CrossRef]
- Ohama, N.; Sato, H.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Transcriptional regulatory network of plant heat stress response. Trends Plant Sci. 2017, 22, 53–65. [Google Scholar] [CrossRef]
- Haider, S.; Raza, A.; Iqbal, J.; Shaukat, M.; Mahmood, T. Analyzing the regulatory role of heat shock transcription factors in plant heat stress tolerance: A brief appraisal. Mol. Biol. Rep. 2022, 49, 5771–5785. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, R.; Song, P.; Zhou, Z. The floral biology, breeding system and pollination efficiency of Schima superba Gardn. et Champ. (Theaceae). Forests 2017, 8, 404. [Google Scholar] [CrossRef]
- Zhang, R.; Yang, H.; Zhou, Z.; Shen, B.; Xiao, J.; Wang, B. A high-density genetic map of Schima superba based on its chromosomal characteristics. BMC Plant Biol. 2019, 19, 41. [Google Scholar] [CrossRef]
- Zheng, W.; Ma, Y.; Tigabu, M.; Yi, Z.; Guo, Y.; Lin, H.; Huang, Z.; Guo, F. Capture of fire smoke particles by leaves of Cunninghamia lanceolata and Schima superba, and importance of leaf characteristics. Sci. Total Environ. 2022, 841, 156772. [Google Scholar] [CrossRef]
- Zhou, G.; Zhou, Y.; Yu, S.; Bai, S.; Lu, F. Schima superba as a fuelbreak: Litter combustibility of three tree species with five water content levels using a cone calorimeter. Front. For. China 2009, 4, 178–184. [Google Scholar] [CrossRef]
- Haider, S.; Iqbal, J.; Naseer, S.; Yaseen, T.; Shaukat, M.; Bibi, H.; Ahmad, Y.; Daud, H.; Abbasi, N.L.; Mahmood, T. Molecular mechanisms of plant tolerance to heat stress: Current landscape and future perspectives. Plant Cell Rep. 2021, 40, 2247–2271. [Google Scholar] [CrossRef]
- Manna, M.; Thakur, T.; Chirom, O.; Mandlik, R.; Deshmukh, R.; Salvi, P. Transcription factors as key molecular target to strengthen the drought stress tolerance in plants. Physiol. Plant. 2021, 172, 847–868. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Song, Z.; Luo, S.; Zhong, Y.; Zhou, Y.; Lu, R. Transcriptome analysis reveals the heat stress response genes by fire stimulation in michelia macclurei dandy. Forests 2023, 14, 610. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef]
- UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2016, 45, D1040–D1045. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, R.; Zhou, Z. Identification and validation of reference genes for gene expression analysis in Schima superba. Genes 2021, 12, 732. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kotak, S.; Larkindale, J.; Lee, U.; von Koskull-Döring, P.; Vierling, E.; Scharf, K.D. Complexity of the heat stress response in plants. Curr. Opin. Plant Biol. 2007, 10, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-F.; Wang, Y.; Tang, Y.; Kakani, V.G.; Mahalingam, R. Transcriptome analysis of heat stress response in switchgrass (Panicum virgatum L.). BMC Plant Biol. 2013, 13, 153. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Gupta, S.K.; Niu, S.; Li, X.Q.; Yang, Q.; Chen, G.; Zhu, W.; Haroon, M. Transcriptome analysis of heat stress response genes in potato leaves. Mol. Biol. Rep. 2020, 47, 4311–4321. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, C.; Zhu, D.; He, H.; Wei, Z.; Yuan, Q.; Li, X.; Gao, X.; Zhang, B.; Gao, H. Identifying candidate genes and patterns of heat-stress response in rice using a genome-wide association study and transcriptome analyses. Crop J. 2022, 10, 1633–1643. [Google Scholar] [CrossRef]
- Usman, M.G.; Rafii, M.; Ismail, M.; Malek, M.; Latif, M.A.; Oladosu, Y. Heat shock proteins: Functions and response against heat stress in plants. Int. J. Sci. Technol. Res. 2014, 3, 204–218. [Google Scholar]
- Feder, M.E.; Hofmann, G.E. Heat-shock proteins, molecular chaperones, and the stress response: Evolutionary and ecological physiology. Annu. Rev. Physiol. 1999, 61, 243–282. [Google Scholar] [CrossRef]
- Tkáčová, J.; Angelovičová, M. Heat shock proteins (HSPs): A review. Sci. Pap. Anim. Sci. Biotechnol. 2012, 45, 349. [Google Scholar]
- Jacob, P.; Hirt, H.; Bendahmane, A. The heat-shock protein/chaperone network and multiple stress resistance. Plant Biotechnol. J. 2017, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Scharf, K.D.; Berberich, T.; Ebersberger, I.; Nover, L. The plant heat stress transcription factor (Hsf) family: Structure, function and evolution. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2012, 1819, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.P.; Sadat, S.; Drenth, J.; McIntyre, C.L. The heat shock factor family from Triticum aestivum in response to heat and other major abiotic stresses and their role in regulation of heat shock protein genes. J. Exp. Bot. 2014, 65, 539–557. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Chen, J.; Liu, Y.; Yang, L.; Li, W.; Zhang, L. Overexpression of Arabidopsis HsfA1a enhances diverse stress tolerance by promoting stress-induced Hsp expression. Genet. Mol. Res. 2014, 13, 1233–1243. [Google Scholar] [CrossRef]
- Jiang, L.; Hu, W.; Qian, Y.; Ren, Q.; Zhang, J. Genome-wide identification, classification and expression analysis of the Hsf and Hsp70 gene families in maize. Gene 2021, 770, 145348. [Google Scholar] [CrossRef]
- Gong, C.; Pang, Q.; Li, Z.; Li, Z.; Chen, R.; Sun, G.; Sun, B. Genome-wide identification and characterization of Hsf and Hsp gene families and gene expression analysis under heat stress in eggplant (Solanum melongema L.). Horticulturae 2021, 7, 149. [Google Scholar] [CrossRef]
- Pucciariello, C.; Banti, V.; Perata, P. ROS signaling as common element in low oxygen and heat stresses. Plant Physiol. Biochem. 2012, 59, 3–10. [Google Scholar] [CrossRef]
- Ara, N.; Nakkanong, K.; Lv, W.; Yang, J.; Hu, Z.; Zhang, M. Antioxidant enzymatic activities and gene expression associated with heat tolerance in the stems and roots of two cucurbit species (“Cucurbita maxima” and “Cucurbita moschata”) and their interspecific inbred line “Maxchata”. Int. J. Mol. Sci. 2013, 14, 24008–24028. [Google Scholar] [CrossRef]
- Chaitanya, K.; Sundar, D.; Masilamani, S.; Ramachandra Reddy, A. Variation in heat stress-induced antioxidant enzyme activities among three mulberry cultivars. Plant Growth Regul. 2002, 36, 175–180. [Google Scholar] [CrossRef]
- Song, A.; Zhu, X.; Chen, F.; Gao, H.; Jiang, J.; Chen, S. A chrysanthemum heat shock protein confers tolerance to abiotic stress. Int. J. Mol. Sci. 2014, 15, 5063–5078. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Xiang, Y. An overview of biomembrane functions in plant responses to high-temperature stress. Front. Plant Sci. 2018, 9, 915. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Huang, Y.; Liu, C.; Chen, K.; Li, M. Functions and interaction of plant lipid signalling under abiotic stresses. Plant Biol. 2023, 25, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, Y.; Huang, B. Lipidomic reprogramming associated with drought stress priming-enhanced heat tolerance in tall fescue (Festuca arundinacea). Plant Cell Environ. 2019, 42, 947–958. [Google Scholar] [CrossRef]
- Ahammed, G.J.; Li, X.; Zhou, J.; Zhou, Y.H.; Yu, J.Q. Role of hormones in plant adaptation to heat stress. In Plant Hormones under Challenging Environmental Factors; Springer: Dordrecht, The Netherlands, 2016; pp. 1–21. [Google Scholar]
- Yoshida, T.; Ohama, N.; Nakajima, J.; Kidokoro, S.; Mizoi, J.; Nakashima, K.; Maruyama, K.; Kim, J.M.; Seki, M.; Todaka, D. Arabidopsis HsfA1 transcription factors function as the main positive regulators in heat shock-responsive gene expression. Mol. Genet. Genom. 2011, 286, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Zhao, Y.; Li, H.; Liu, W. Wheat heat shock factor TaHsfA6f increases ABA levels and enhances tolerance to multiple abiotic stresses in transgenic plants. Int. J. Mol. Sci. 2020, 21, 3121. [Google Scholar] [CrossRef]
- Toh, S.; Imamura, A.; Watanabe, A.; Nakabayashi, K.; Okamoto, M.; Jikumaru, Y.; Hanada, A.; Aso, Y.; Ishiyama, K.; Tamura, N. High temperature-induced abscisic acid biosynthesis and its role in the inhibition of gibberellin action in Arabidopsis seeds. Plant Physiol. 2008, 146, 1368–1385. [Google Scholar] [CrossRef]
- Sakata, T.; Oshino, T.; Miura, S.; Tomabechi, M.; Tsunaga, Y.; Higashitani, N.; Miyazawa, Y.; Takahashi, H.; Watanabe, M.; Higashitani, A. Auxins reverse plant male sterility caused by high temperatures. Proc. Natl. Acad. Sci. USA 2010, 107, 8569–8574. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Guan, H.; Li, B.; Zhang, S.; Xu, Y.; Yao, Y.; Yang, X.; Zha, Z.; Guo, Y.; Jiao, C. Transcriptome analysis reveals the dynamic and rapid transcriptional reprogramming involved in heat stress and identification of heat response genes in rice. Int. J. Mol. Sci. 2023, 24, 14802. [Google Scholar] [CrossRef]
- Cheng, M.C.; Liao, P.M.; Kuo, W.W.; Lin, T.P. The Arabidopsis ETHYLENE RESPONSE FACTOR1 regulates abiotic stress-responsive gene expression by binding to different cis-acting elements in response to different stress signals. Plant Physiol. 2013, 162, 1566–1582. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhao, X.; Bürger, M.; Chory, J.; Wang, X. The role of ethylene in plant temperature stress response. Trends Plant Sci. 2023, 28, 808–824. [Google Scholar] [CrossRef]
- Wu, Y.; Su, S.; Wang, T.; Peng, G.H.; He, L.; Long, C.; Li, W. Identification and expression characteristics of NLP (NIN-like protein) gene family in pepper (Capsicum annuum L.). Mol. Biol. Rep. 2023, 50, 6655–6668. [Google Scholar] [CrossRef]
- von Koskull-Döring, P.; Scharf, K.-D.; Nover, L. The diversity of plant heat stress transcription factors. Trends Plant Sci. 2007, 12, 452–457. [Google Scholar] [CrossRef]
- Song, A.; Li, P.; Jiang, J.; Chen, S.; Li, H.; Zeng, J.; Shao, Y.; Zhu, L.; Zhang, Z.; Chen, F. Phylogenetic and transcription analysis of Chrysanthemum WRKY transcription factors. Int. J. Mol. Sci. 2014, 15, 14442–14455. [Google Scholar] [CrossRef]
- Jiang, Y.; Jiang, D.; Xia, M.; Gong, M.; Li, H.; Xing, H.; Zhu, X.; Li, H.L. Genome-wide identification and expression analysis of the TCP gene family related to developmental and abiotic stress in ginger. Plants 2023, 12, 3389. [Google Scholar] [CrossRef]
- Chanwala, J.; Khadanga, B.; Jha, D.K.; Sandeep, I.S.; Dey, N. MYB transcription factor family in pearl millet: Genome-wide identification, evolutionary progression and expression analysis under abiotic stress and phytohormone treatments. Plants 2023, 12, 355. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item. | Count | Percentage |
|---|---|---|
| Nr | 34,006 | 21.19% |
| Uniprot | 33,691 | 21.00% |
| GO | 24,105 | 15.02% |
| KEGG | 20,238 | 12.61% |
| Pfam | 16,631 | 10.37% |
| Pathway | 10,068 | 6.27% |
| KOG | 195 | 0.12% |
| Annotation | 34,308 | 21.38% |
| All | 160,449 | 100.00% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, S.; Zhong, Y.; Wen, W.; Yu, C.; Lu, R.; Luo, S. Transcriptome Analysis Identifies Key Genes Involved in Response and Recovery to High Heat Stress Induced by Fire in Schima superba. Genes 2024, 15, 1108. https://doi.org/10.3390/genes15081108
Wei S, Zhong Y, Wen W, Yu C, Lu R, Luo S. Transcriptome Analysis Identifies Key Genes Involved in Response and Recovery to High Heat Stress Induced by Fire in Schima superba. Genes. 2024; 15(8):1108. https://doi.org/10.3390/genes15081108
Chicago/Turabian StyleWei, Shujing, Yingxia Zhong, Wen Wen, Chong Yu, Ruisen Lu, and Sisheng Luo. 2024. "Transcriptome Analysis Identifies Key Genes Involved in Response and Recovery to High Heat Stress Induced by Fire in Schima superba" Genes 15, no. 8: 1108. https://doi.org/10.3390/genes15081108
APA StyleWei, S., Zhong, Y., Wen, W., Yu, C., Lu, R., & Luo, S. (2024). Transcriptome Analysis Identifies Key Genes Involved in Response and Recovery to High Heat Stress Induced by Fire in Schima superba. Genes, 15(8), 1108. https://doi.org/10.3390/genes15081108