
Revealing the Genetic Diversity and Population Structure of Garlic Resource Cultivars and Screening of Core Cultivars Based on Specific Length Amplified Fragment Sequencing (SLAF-Seq)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Preparation and Resequencing of DNA Libraries
2.3. Data Analysis
2.4. Population Genetic Analysis
2.5. Core Germplasm Screening
2.6. Analysis of Genetic Diversity in the Core Germplasm
3. Results
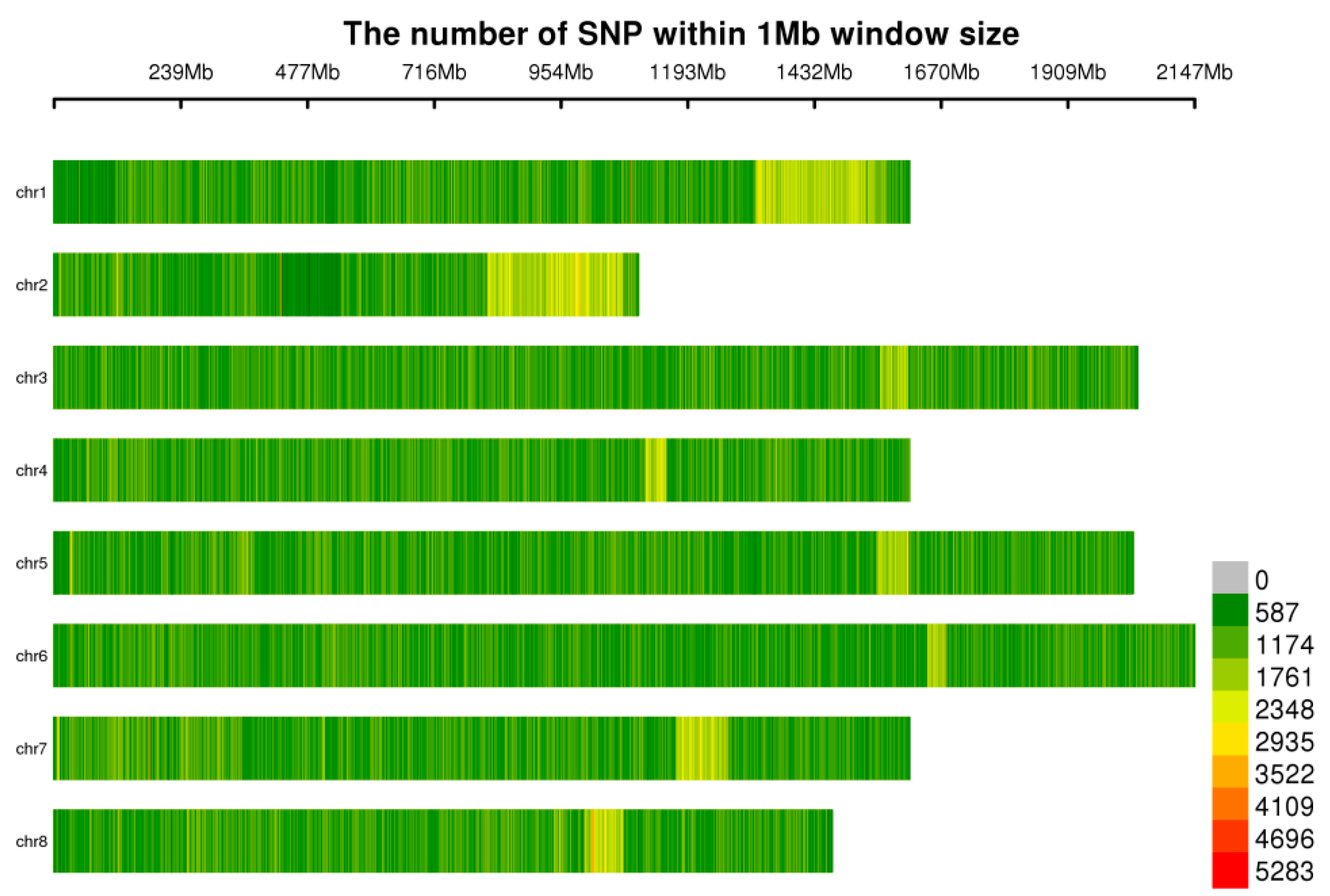
3.1. Assessment of the Quality of the Resequencing Data from 102 Garlic Genomes
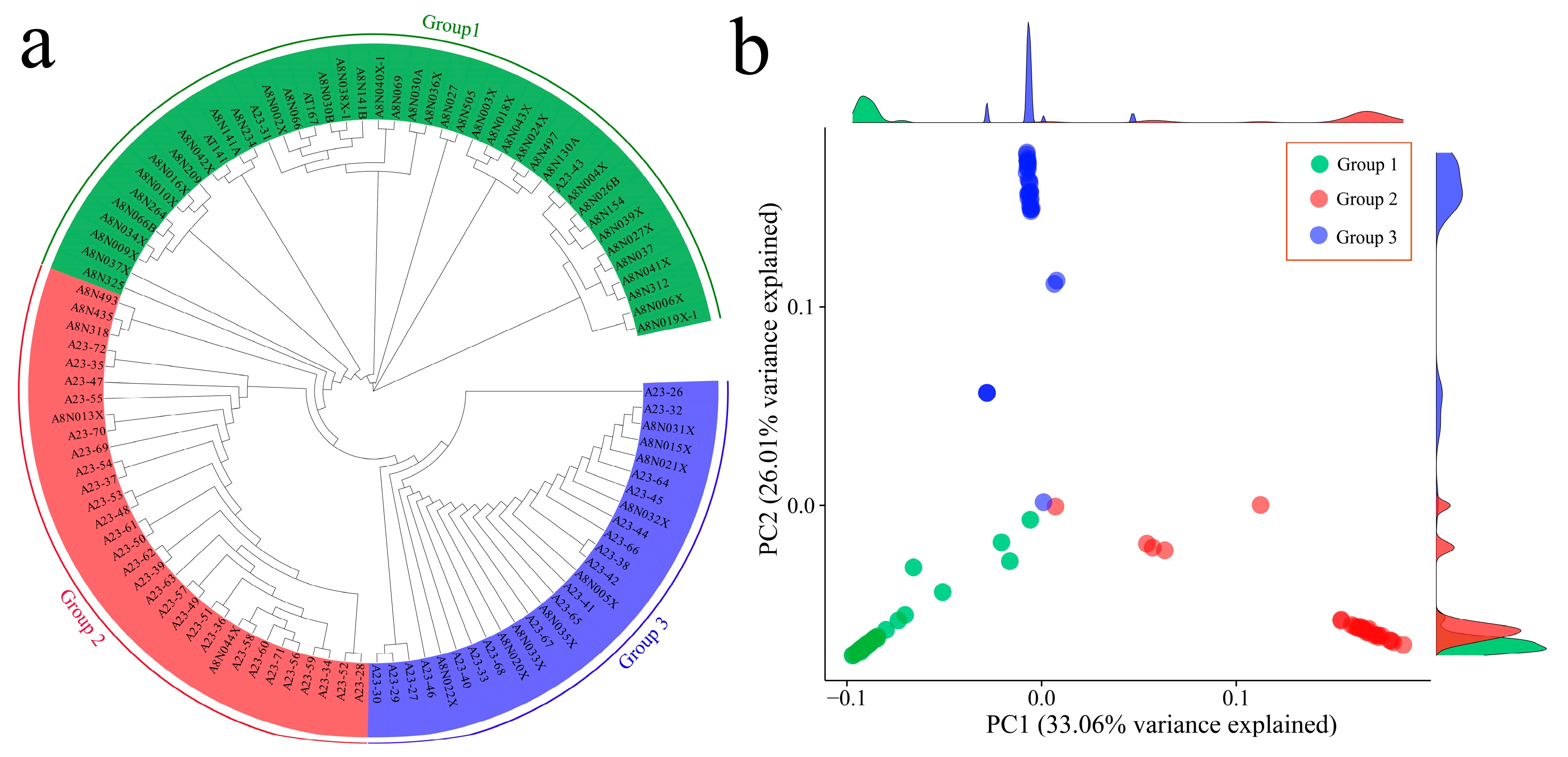
3.2. Phylogenetic and Principal Component Analyses of 102 Garlic Genomes
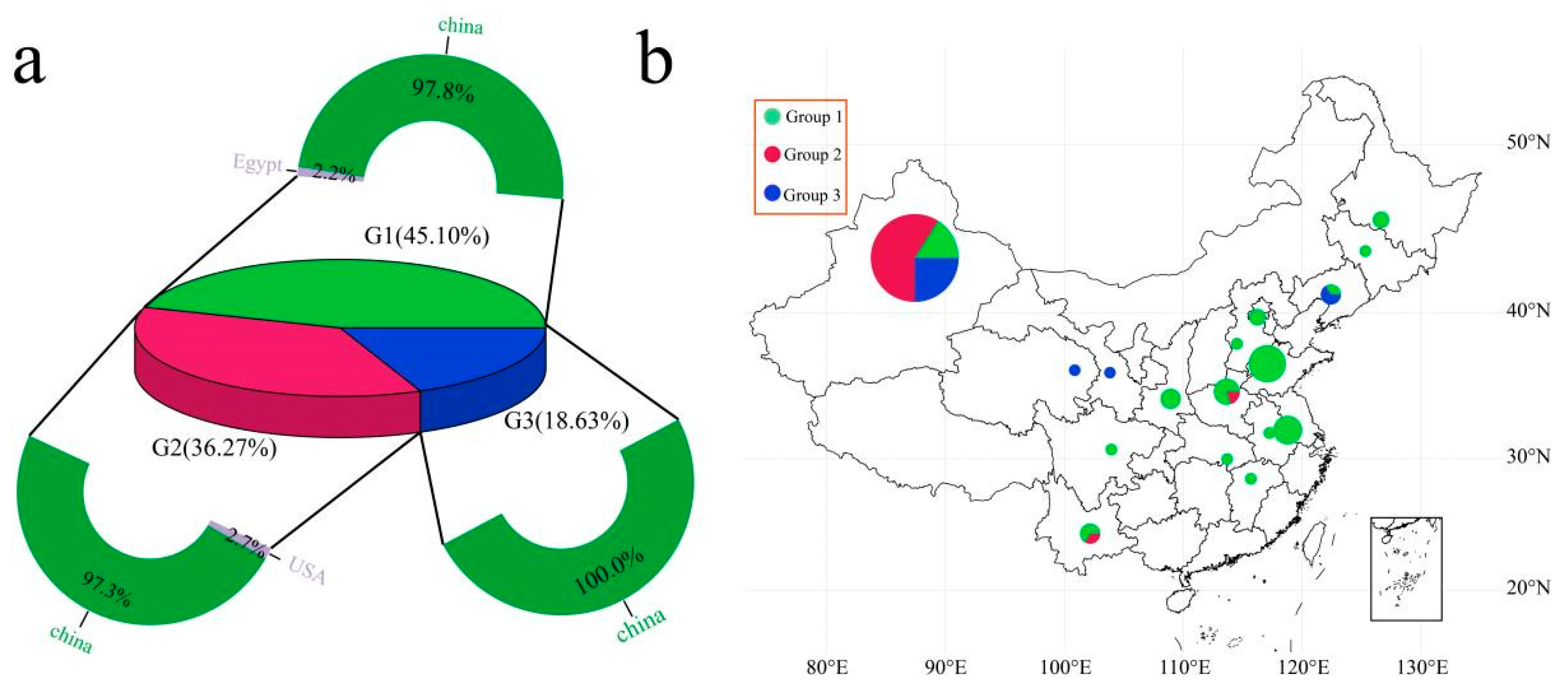
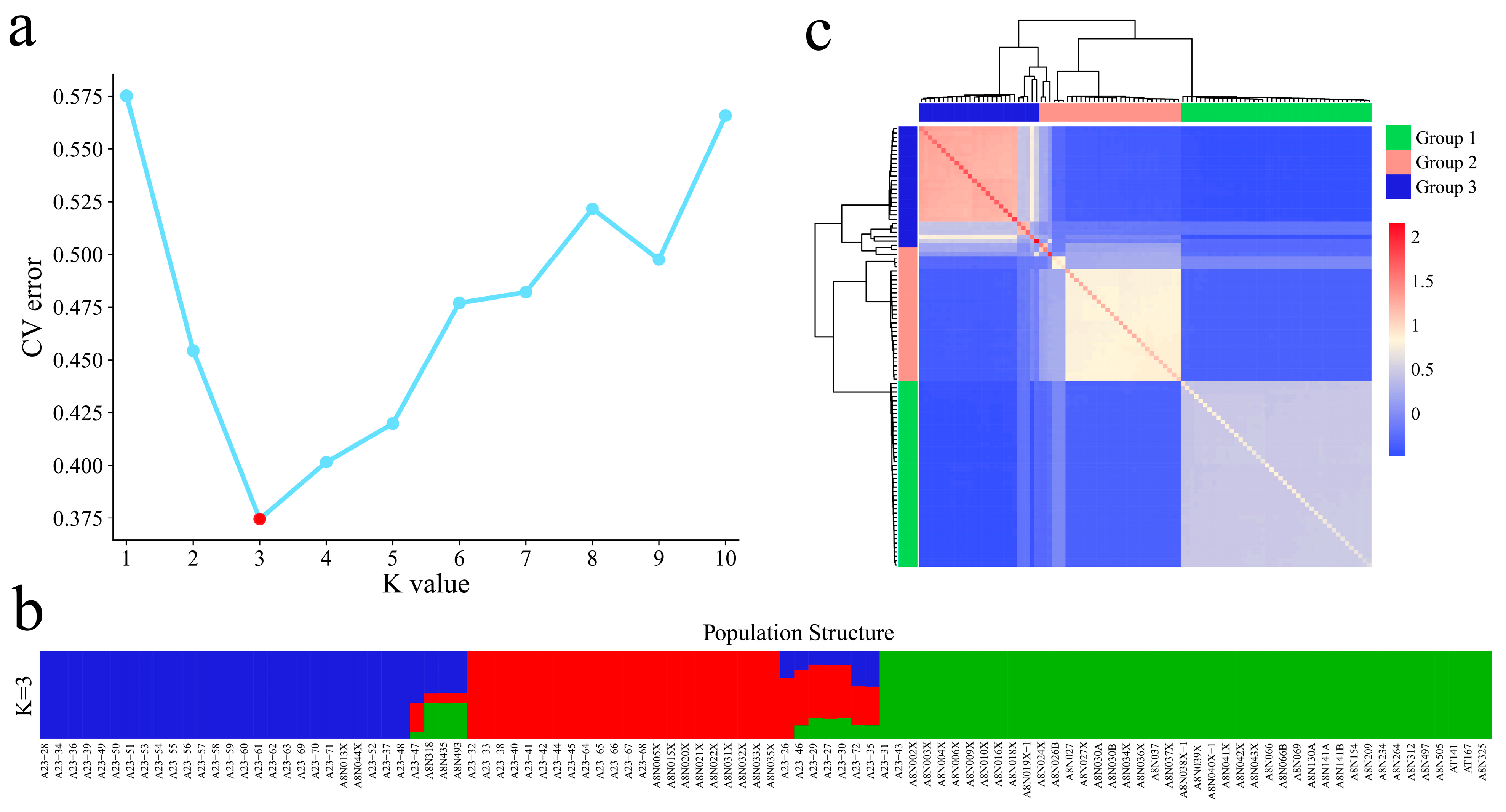
3.3. Population Structure and Genetic Relationship Analyses of 102 Garlic Genomes
3.4. Analysis of the Genetic Diversity between Groups
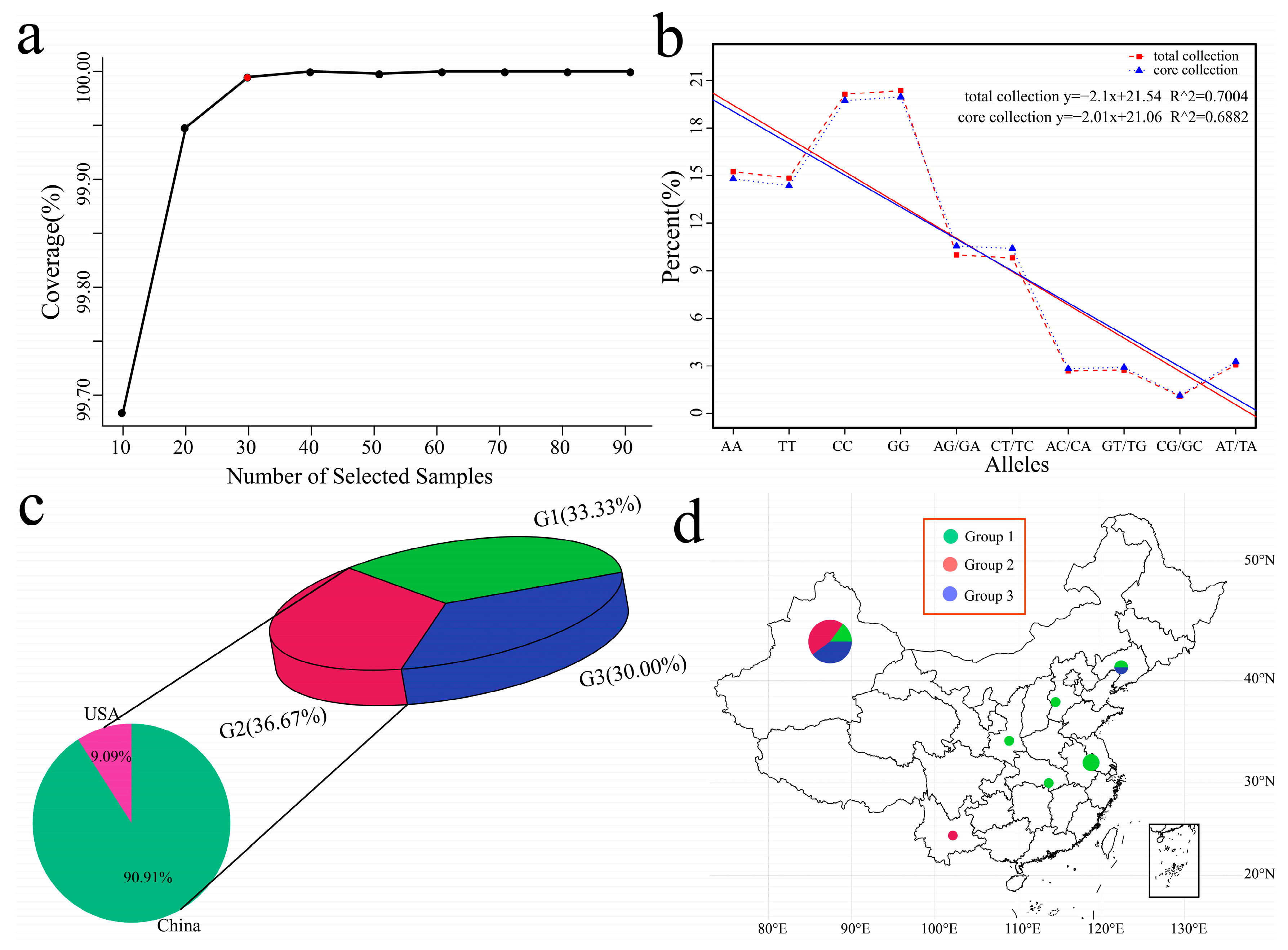
3.5. Core Material Screening
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, X.; Zhu, S.; Li, N.; Cheng, Y.; Zhao, J.; Qiao, X.; Lu, L.; Liu, S.; Wang, Y.; Liu, C.; et al. A Chromosome-Level Genome Assembly of Garlic (Allium sativum) Provides Insights into Genome Evolution and Allicin Biosynthesis. Mol. Plant 2020, 13, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Chidike Ezeorba, T.P.; Ezugwu, A.L.; Chukwuma, I.F.; Anaduaka, E.G.; Udenigwe, C.C. Health-promoting properties of bioactive proteins and peptides of garlic (Allium sativum). Food Chem. 2024, 435, 137632. [Google Scholar] [CrossRef]
- Fritsch, R.M.; Friesen, N. Chapter 1: Evolution, Domestication, and Taxonomy. In Allium Crop Science: Recent Advances; Rabinowitch, H.D., Currah, L., Eds.; CABI Publishing: Wallingford, UK, 2002; Volume 19. [Google Scholar]
- Li, N.; Zhang, X.; Sun, X.; Zhu, S.; Cheng, Y.; Liu, M.; Gao, S.; Zhang, J.; Wang, Y.; Yang, X.; et al. Genomic insights into the evolutionary history and diversification of bulb traits in garlic. Genome Biol. 2022, 23, 188. [Google Scholar] [CrossRef] [PubMed]
- Stępień, A.E.; Trojniak, J.; Tabarkiewicz, J. Anti-Cancer Anti-Inflammatory Properties of Black Garlic. Int. J. Mol. Sci. 2024, 25, 1801. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.J.; Lee, A.; Min, S.S.; In, S.; Kim, E.; Kim, H.J. Comparison of Physicochemical Characteristics of Garlic Produced from South Korea and China. J. Food Sci. 2019, 84, 1806–1811. [Google Scholar] [CrossRef] [PubMed]
- Luebert, F.; Scherson, R.A. Choice of molecular marker influences spatial patterns of phylogenetic diversity. Biol. Lett. 2024, 20, 20230581. [Google Scholar] [CrossRef]
- Maaß, H.I.; Klaas, M. Infraspecific differentiation of garlic (Allium sativum L.) by isozyme and RAPD markers. TAG. Theoretical and applied genetics. Theor. Appl. Genet. 1995, 91, 89–97. [Google Scholar] [CrossRef]
- Bradley, K.F.; Rieger, M.A.; Collins, G.G. Classification of Australian garlic cultivars by DNA fingerprinting. Aust. J. Exp. Agric. 1996, 36, 613–618. [Google Scholar] [CrossRef]
- Al-Zahim, M.A.; Ford-Lloyd, B.V.; Newbury, H.J. Detection of somaclonal variation in garlic (Allium sativum L.) using RAPD and cytological analysis. Plant Cell Rep. 1999, 18, 473–477. [Google Scholar] [CrossRef]
- Ipek, M.; Simon, P. Genetic Diversity in Garlic (Allium sativum L.) as assessed by AFLPs and Isozymes. In Proceedings of the American Society for Horticultural Science 98th Annual Conference & Exhibition, Sacramento, CA, USA, 22–25 July 2001. [Google Scholar]
- García Lampasona, S.; Martinez, L.; Burba, J.L. Genetic diversity among selected Argentinean garlic clones (Allium sativum L.) using AFLP (Amplified Fragment Length Polymorphism). Euphytica 2003, 132, 115–119. [Google Scholar] [CrossRef]
- Volk, G.M.; Henk, A.D.; Richards, C.M. Genetic diversity among US garlic clones as detected using AFLP methods. J. Am. Soc. Hortic. Sci. 2004, 129, 559–569. [Google Scholar] [CrossRef]
- Ipek, M.; Ipek, A.; Almquist, S.G.; Simon, P.W. Demonstration of linkage and development of the first low-density genetic map of garlic, based on AFLP markers. TAG. Theoretical and applied genetics. Theor. Appl. Genet. 2005, 110, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Ovesná, J.; Kučera, L.; Horníčková, J.; Svobodová, L.; Stavělíková, H.; Velíšek, J.; Milella, L. Diversity of S-alk (en) yl cysteine sulphoxide content within a collection of garlic (Allium sativum L.) and its association with the morphological and genetic background assessed by AFLP. Sci. Hortic. 2011, 129, 541–547. [Google Scholar] [CrossRef]
- Kamenetsky, R.; Faigenboim, A.; Shemesh Mayer, E.; Ben Michael, T.; Gershberg, C.; Kimhi, S.; Esquira, I.; Rohkin Shalom, S.; Eshel, D.; Rabinowitch, H.D.; et al. Integrated transcriptome catalogue and organ-specific profiling of gene expression in fertile garlic (Allium sativum L.). BMC Genom. 2015, 16, 12. [Google Scholar] [CrossRef]
- Jabbes, N.; Geoffriau, E.E.; Le Clerc, V.; Dridi, B.; Hannechi, C. Inter simple sequence repeat fingerprints for assess genetic diversity of Tunisian garlic populations. J. Agric. Sci. 2011, 3, 77–85. [Google Scholar] [CrossRef]
- Son, J.H.; Park, K.C.; Lee, S.I.; Kim, J.H.; Kim, N.S. Species relationships among Allium species by ISSR analysis. Hortic. Environ. Biotechnol. 2012, 53, 256–262. [Google Scholar] [CrossRef]
- Mandlik, J.S.; Patil, A.S.; Singh, S. Next-Generation Sequencing (NGS): Platforms and Applications. J. Pharm. Bioallied Sci. 2024, 16, S41–S45. [Google Scholar] [CrossRef]
- Preston, J.C. Insights into the evo-devo of plant reproduction using next-generation sequencing approaches. J. Exp. Bot. 2021, 72, 1536–1545. [Google Scholar] [CrossRef] [PubMed]
- Boatwright, J.L.; Sapkota, S.; Jin, H.; Schnable, J.C.; Brenton, Z.; Boyles, R.; Kresovich, S. Sorghum Association Panel whole-genome sequencing establishes cornerstone resource for dissecting genomic diversity. Plant J. 2022, 111, 888–904. [Google Scholar] [CrossRef]
- Tomar, V.; Dhillon, G.S.; Singh, D.; Singh, R.P.; Poland, J.; Joshi, A.K.; Tiwari, B.S.; Kumar, U. Elucidating SNP-based genetic diversity and population structure of advanced breeding lines of bread wheat (Triticum aestivum L.). PeerJ 2021, 9, e11593. [Google Scholar] [CrossRef]
- Xie, P.J.; Ke, Y.T.; Kuo, L.Y. Modified CTAB protocols for high-molecular-weight DNA extractions from ferns. Appl. Plant Sci. 2023, 11, e11526. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Betschart, R.O.; Thiéry, A.; Aguilera-Garcia, D.; Zoche, M.; Moch, H.; Twerenbold, R.; Zeller, T.; Blankenberg, S.; Ziegler, A. Comparison of calling pipelines for whole genome sequencing: An empirical study demonstrating the importance of mapping and alignment. Sci. Rep. 2022, 12, 21502. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Marth, G.; Abecasis, G.; Durbin, R.; et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Montana, G.; Hoggart, C. Statistical software for gene mapping by admixture linkage disequilibrium. Brief. Bioinform. 2007, 8, 393–395. [Google Scholar] [CrossRef]
- Francis, R.M. pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- De Beukelaer, H.; Smýkal, P.; Davenport, G.F.; Fack, V. Core Hunter II: Fast core subset selection based on multiple genetic diversity measures using Mixed Replica search. BMC Bioinform. 2012, 13, 312. [Google Scholar] [CrossRef]
- Dolferus, R.; Onyemaobi, O. Editorial on Genetic Diversity of Plant Tolerance to Environmental Restraints. Genes 2023, 14, 1992. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Wei, Z.; Zhou, B.; Chen, X.; Gao, Q.; Zhang, Z. Molecular marker development and genetic diversity exploration in Medicago polymorpha. PeerJ 2023, 11, e14698. [Google Scholar] [CrossRef] [PubMed]
- Nazari, M.; Tohidfar, M.; Ramshini, H.; Vahdati, K. Molecular and morphological evaluation of transgenic Persian walnut plants harboring Fld gene under osmotic stress condition. Mol. Biol. Rep. 2022, 49, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Tuvesson, S.D.; Larsson, C.T.; Ordon, F. Use of Molecular Markers for Doubled Haploid Technology: From Academia to Plant Breeding Companies. Methods Mol. Biol. 2021, 2288, 49–72. [Google Scholar] [CrossRef]
- Sun, Y.; Shang, L.; Zhu, Q.H.; Fan, L.; Guo, L. Twenty years of plant genome sequencing: Achievements and challenges. Trends Plant Sci. 2022, 27, 391–401. [Google Scholar] [CrossRef]
- Shi, J.; Tian, Z.; Lai, J.; Huang, X. Plant pan-genomics and its applications. Mol. Plant 2023, 16, 168–186. [Google Scholar] [CrossRef]
- Zia, M.A.B.; Yousaf, M.F.; Asim, A.; Naeem, M. An overview of genome-wide association mapping studies in Poaceae species (model crops: Wheat and rice). Mol. Biol. Rep. 2022, 49, 12077–12090. [Google Scholar] [CrossRef]
- Bal, S.; Sharangi, A.B.; Upadhyay, T.K.; Aljerwan, R.F.; Altayyar, A.A.; Kahrizi, D.; Ahmad, I. Quantum leap in the light of molecular elucidation of garlic genome. Cell. Mol. Biol. 2023, 69, 33–51. [Google Scholar] [CrossRef]
- Yang, C.; Yan, J.; Jiang, S.; Li, X.; Min, H.; Wang, X.; Hao, D. Resequencing 250 Soybean Accessions: New Insights into Genes Associated with Agronomic Traits and Genetic Networks. Genom. Proteom. Bioinform. 2022, 20, 29–41. [Google Scholar] [CrossRef]
- Grzybowski, M.W.; Mural, R.V.; Xu, G.; Turkus, J.; Yang, J.; Schnable, J.C. A common resequencing-based genetic marker data set for global maize diversity. Plant J. 2023, 113, 1109–1121. [Google Scholar] [CrossRef]
- Zhang, L.; Deng, B.; Peng, Y.; Gao, Y.; Hu, Y.; Bao, J. Population Structure and Genetic Diversity of Shanlan Landrace Rice for GWAS of Cooking and Eating Quality Traits. Int. J. Mol. Sci. 2024, 25, 3469. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Sun, M.; Ren, X.; Li, P.; Hui, J.; Zhang, J.; Lin, G. Revealing the Genetic Diversity and Population Structure of Garlic Resource Cultivars and Screening of Core Cultivars Based on Specific Length Amplified Fragment Sequencing (SLAF-Seq). Genes 2024, 15, 1135. https://doi.org/10.3390/genes15091135
Yang J, Sun M, Ren X, Li P, Hui J, Zhang J, Lin G. Revealing the Genetic Diversity and Population Structure of Garlic Resource Cultivars and Screening of Core Cultivars Based on Specific Length Amplified Fragment Sequencing (SLAF-Seq). Genes. 2024; 15(9):1135. https://doi.org/10.3390/genes15091135
Chicago/Turabian StyleYang, Jing, Meile Sun, Xiangrong Ren, Pengbing Li, Jingtao Hui, Jun Zhang, and Guocang Lin. 2024. "Revealing the Genetic Diversity and Population Structure of Garlic Resource Cultivars and Screening of Core Cultivars Based on Specific Length Amplified Fragment Sequencing (SLAF-Seq)" Genes 15, no. 9: 1135. https://doi.org/10.3390/genes15091135
APA StyleYang, J., Sun, M., Ren, X., Li, P., Hui, J., Zhang, J., & Lin, G. (2024). Revealing the Genetic Diversity and Population Structure of Garlic Resource Cultivars and Screening of Core Cultivars Based on Specific Length Amplified Fragment Sequencing (SLAF-Seq). Genes, 15(9), 1135. https://doi.org/10.3390/genes15091135