
In Silico Analysis of the Missense Variants of Uncertain Significance of CTNNB1 Gene Reported in GnomAD Database

 ,
,  , , and
, , and
Abstract
:1. Introduction
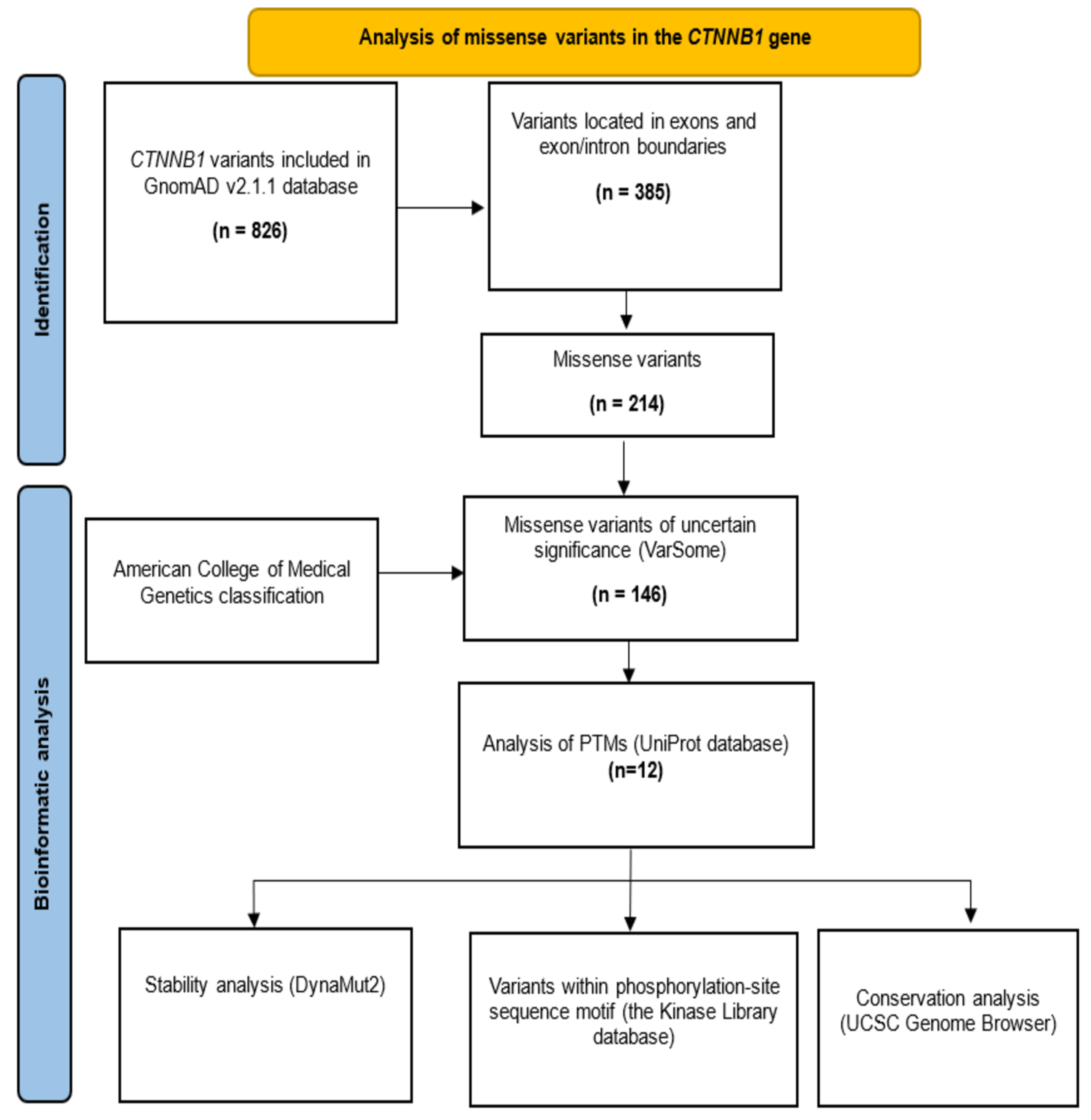
2. Materials and Methods
2.1. Selection of Missense VUS
2.2. Impact of Missense VUS on Phosphorylation Sites
2.3. Conservation Analysis of Affected Residues
2.4. Stability Analysis of Missense VUS
3. Results
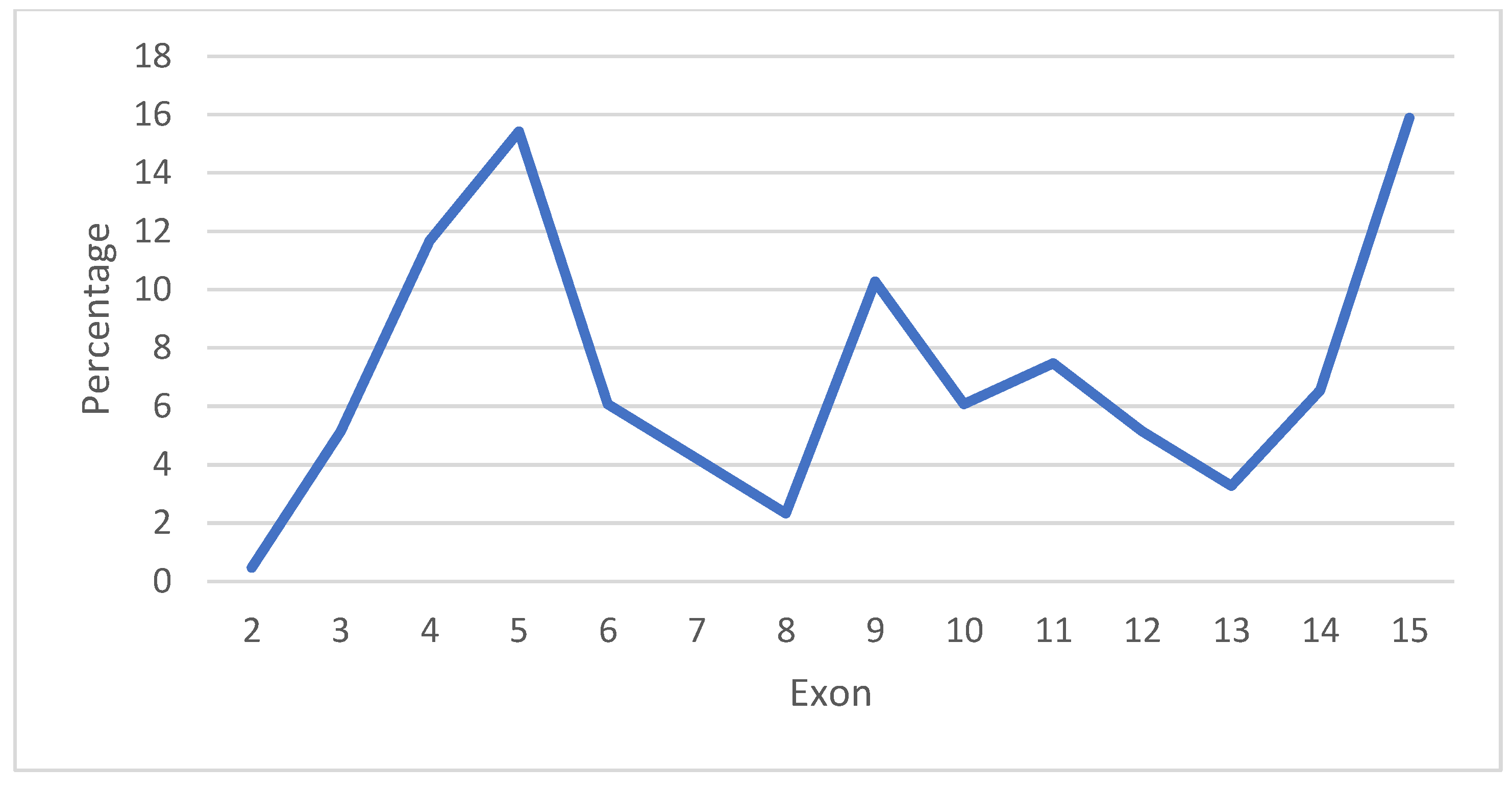
3.1. Analysis of Variants of the CTNNB1 Gene
3.2. In Silico Analysis of Phosphorylation Motif Sequence
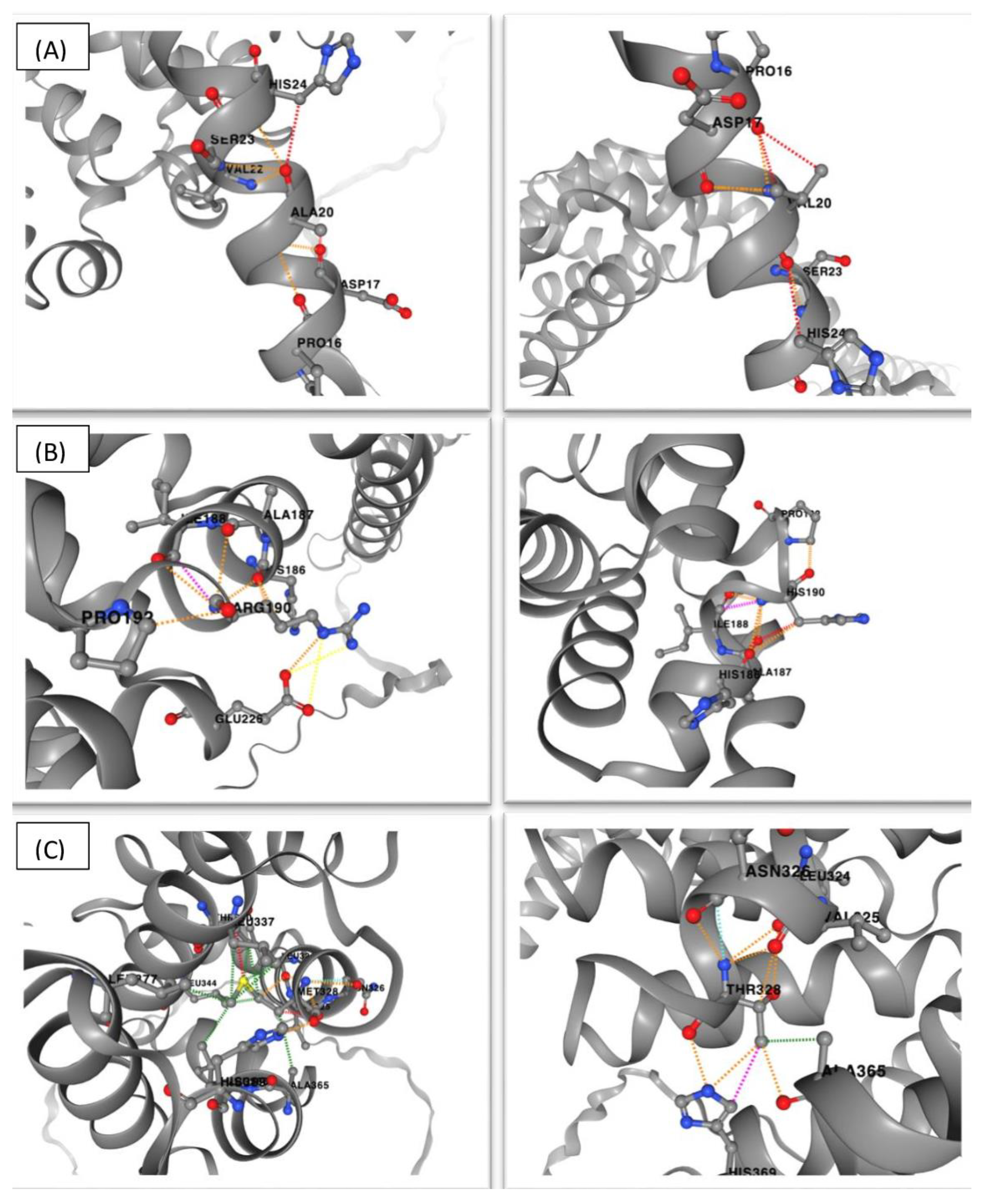
3.3. Impact on and Changes in the Protein Structure Analysis
3.4. Conservation Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sayers, E.W.; Beck, J.; Brister, J.R.; Bolton, E.E.; Canese, K.; Comeau, D.C.; Funk, K.; Ketter, A.; Kim, S.; Kimchi, A.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2020, 48, D9–D16. [Google Scholar] [CrossRef]
- Kim, S.; Jeong, S. Mutation hotspots in the β-catenin gene: Lessons from the human cancer genome databases. Mol. Cells 2019, 42, 8–16. [Google Scholar] [CrossRef]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef]
- Zhuang, W.; Ye, T.; Wang, W.; Song, W.; Tan, T. CTNNB1 in neurodevelopmental disorders. Front. Psychiatry 2023, 14, 1143328. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Johnson, J.L.; Yaron, T.M.; Huntsman, E.M.; Kerelsky, A.; Song, J.; Regev, A.; Lin, T.Y.; Liberatore, K.; Cizin, D.M.; Cohen, B.M.; et al. An atlas of substrate specificities for the human serine/threonine kinome. Nature 2023, 613, 759–766. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Rodrigues, C.H.M.; Pires, D.E.V.; Ascher, D.B. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 2021, 30, 60–69. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Corso, G.; Corso, F.; Bellerba, F.; Carneiro, P.; Seixas, S.; Cioffi, A.; La Vecchia, C.; Magnoni, F.; Bonanni, B.; Veronesi, P.; et al. Geographical distribution of e-cadherin germline mutations in the context of diffuse gastric cancer: A systematic review. Cancers 2021, 13, 1269. [Google Scholar] [CrossRef]
- Ha, J.R.; Hao, L.; Venkateswaran, G.; Huang, Y.H.; Garcia, E.; Persad, S. β-Catenin is O-GlcNAc glycosylated at Serine 23: Implications for β-catenin’s subcellular localization and transactivator function. Exp. Cell Res. 2014, 321, 153–166. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Vasovcak, P.; Pavlikova, K.; Sedlacek, Z.; Skapa, P.; Kouda, M.; Hoch, J.; Krepelova, A. Molecular genetic analysis of 103 sporadic colorectal tumours in Czech patients. PLoS ONE 2011, 6, e24114. [Google Scholar] [CrossRef]
- Stamos, J.L.; Weis, W.I. The β-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef]
- Campillo-Marcos, I.; García-González, R.; Navarro-Carrasco, E.; Lazo, P.A. The human VRK1 chromatin kinase in cancer biology. Cancer Lett. 2021, 503, 117–128. [Google Scholar] [CrossRef]
- Hardman, G.; Perkins, S.; Brownridge, P.J.; Clarke, C.J.; Byrne, D.P.; Campbell, A.E.; Kalyuzhnyy, A.; Myall, A.; Eyers, P.A.; Jones, A.R.; et al. Strong anion exchange-mediated phosphoproteomics reveals extensive human non-canonical phosphorylation. EMBO J. 2019, 38, e100847. [Google Scholar] [CrossRef] [PubMed]
- Esmaili, F.; Pourmirzaei, M.; Ramazi, S.; Shojaeilangari, S.; Yavari, E. A Review of Machine Learning and Algorithmic Methods for Protein Phosphorylation Site Prediction. Genom. Proteom. Bioinform. 2023, 21, 1266–1285. [Google Scholar] [CrossRef] [PubMed]
- Kurniawan, A.N.; Hongyo, T.; Hardjolukito, E.S.; Ham, M.F.; Takakuwa, T.; Kodariah, R.; Hoshida, Y.; Nomura, T.; Aozasa, K. Gene mutation analysis of sinonasal lymphomas in Indonesia. Oncol. Rep. 2006, 15, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 7, Unit7.20. [Google Scholar] [CrossRef] [PubMed]
- Van der Wal, T.; van Amerongen, R. Walking the tight wire between cell adhesion and WNT signalling: A balancing act for β-catenin. Open Biol. 2020, 10, 200267. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Wang, Y.; Broaddus, R.; Sun, L.; Xue, F.; Zhang, W. Exon 3 mutations of CTNNB1 drive tumorigenesis: A review. Oncotarget 2017, 9, 5492–5508. [Google Scholar] [CrossRef]
- Kitamura, N.; Galligan, J.J. A global view of the human post-translational modification landscape. Biochem. J. 2023, 480, 1241–1265. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTNNB1 Variant Type | N (%) |
|---|---|
| Missense | 214 (56) |
| Synonymous | 167 (43) |
| Others | 4 (1) |
| ACMG Classification | N (%) |
|---|---|
| Likely pathogenic | 3 (1) |
| VUS | 146 (68) |
| Likely benign | 54 (25) |
| Benign | 11 (5) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caballero-Avendaño, A.; Gutiérrez-Angulo, M.; Ayala-Madrigal, M.d.l.L.; Moreno-Ortiz, J.M.; González-Mercado, A.; Peregrina-Sandoval, J. In Silico Analysis of the Missense Variants of Uncertain Significance of CTNNB1 Gene Reported in GnomAD Database. Genes 2024, 15, 972. https://doi.org/10.3390/genes15080972
Caballero-Avendaño A, Gutiérrez-Angulo M, Ayala-Madrigal MdlL, Moreno-Ortiz JM, González-Mercado A, Peregrina-Sandoval J. In Silico Analysis of the Missense Variants of Uncertain Significance of CTNNB1 Gene Reported in GnomAD Database. Genes. 2024; 15(8):972. https://doi.org/10.3390/genes15080972
Chicago/Turabian StyleCaballero-Avendaño, Arturo, Melva Gutiérrez-Angulo, María de la Luz Ayala-Madrigal, José Miguel Moreno-Ortiz, Anahí González-Mercado, and Jorge Peregrina-Sandoval. 2024. "In Silico Analysis of the Missense Variants of Uncertain Significance of CTNNB1 Gene Reported in GnomAD Database" Genes 15, no. 8: 972. https://doi.org/10.3390/genes15080972