
Integrating Multi-Omics Data to Identify Key Functional Variants Affecting Feed Efficiency in Large White Boars
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Muscle and Duodenal Tissue Samples
2.2. Construction of RNA-Seq Libraries
2.3. Construction of ATAC-Seq Libraries
2.4. Collection of Animal Population Data, Phenotypic Data, and Genotyping
2.5. Extreme Phenotype Selection in Population Subgroups
2.6. RNA-Seq Data Analysis
2.7. ATAC-Seq Data Analysis
2.8. Gene Function Annotation
2.9. Genotyping and Quality Control
2.10. Genotype Imputation and Accuracy Assessment
2.11. GWAS
3. Results
3.1. Mapping Quantitative Trait Loci for Pig Feed Efficiency
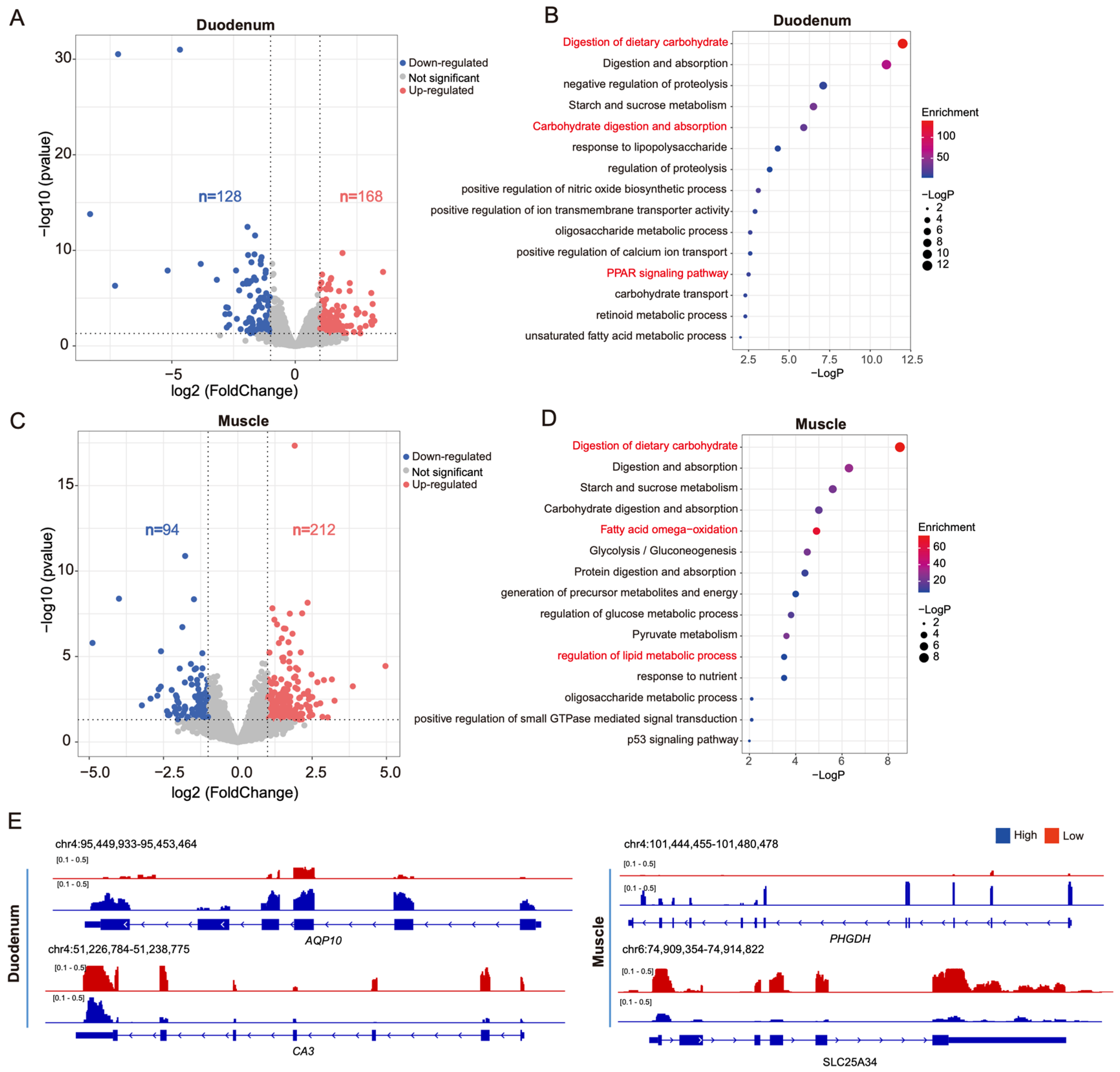
3.2. Differential Gene Transcription in Pigs with High and Low Feed Efficiency
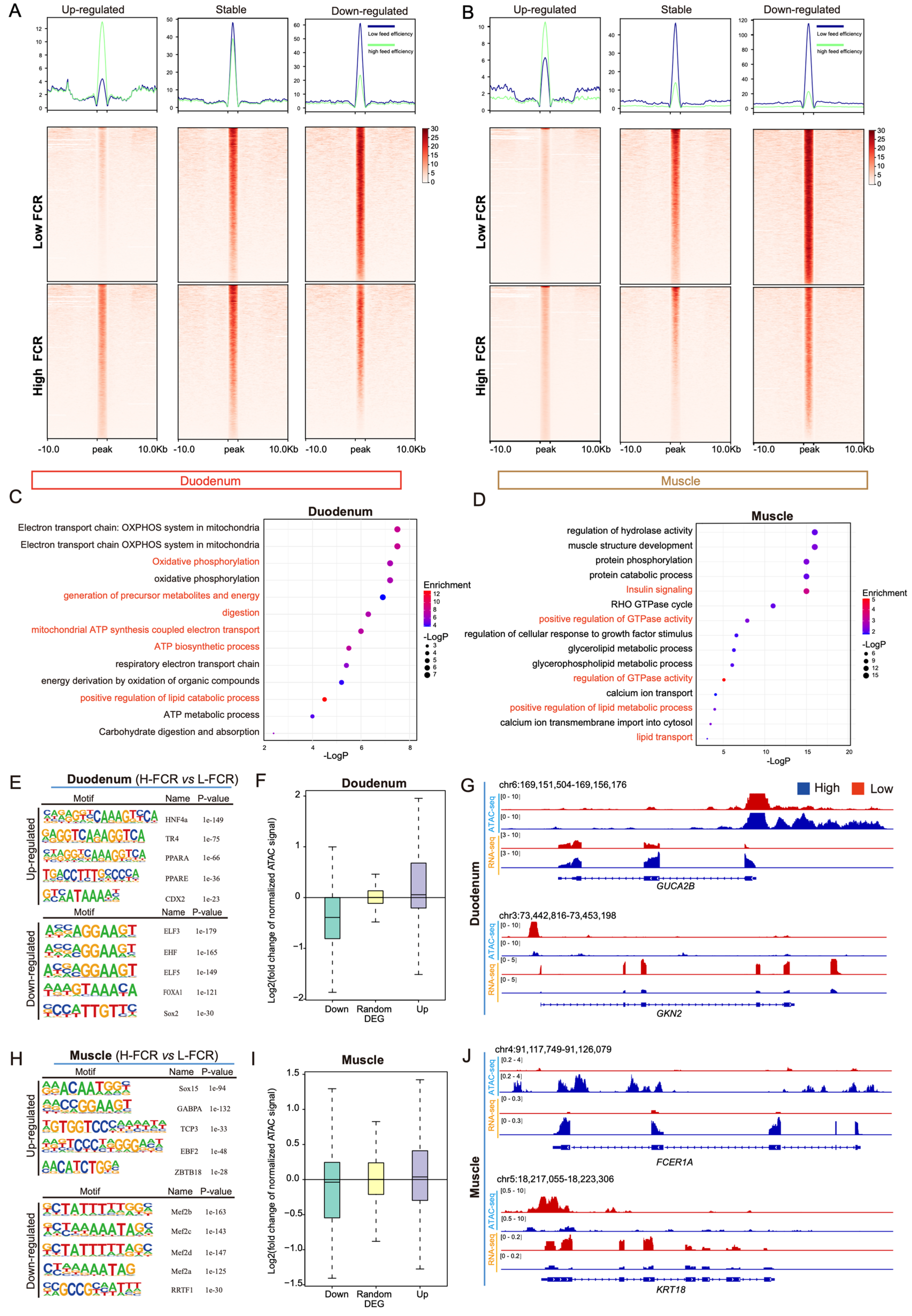
3.3. Differences in Chromatin Accessibility between High- and Low-FCR Pigs
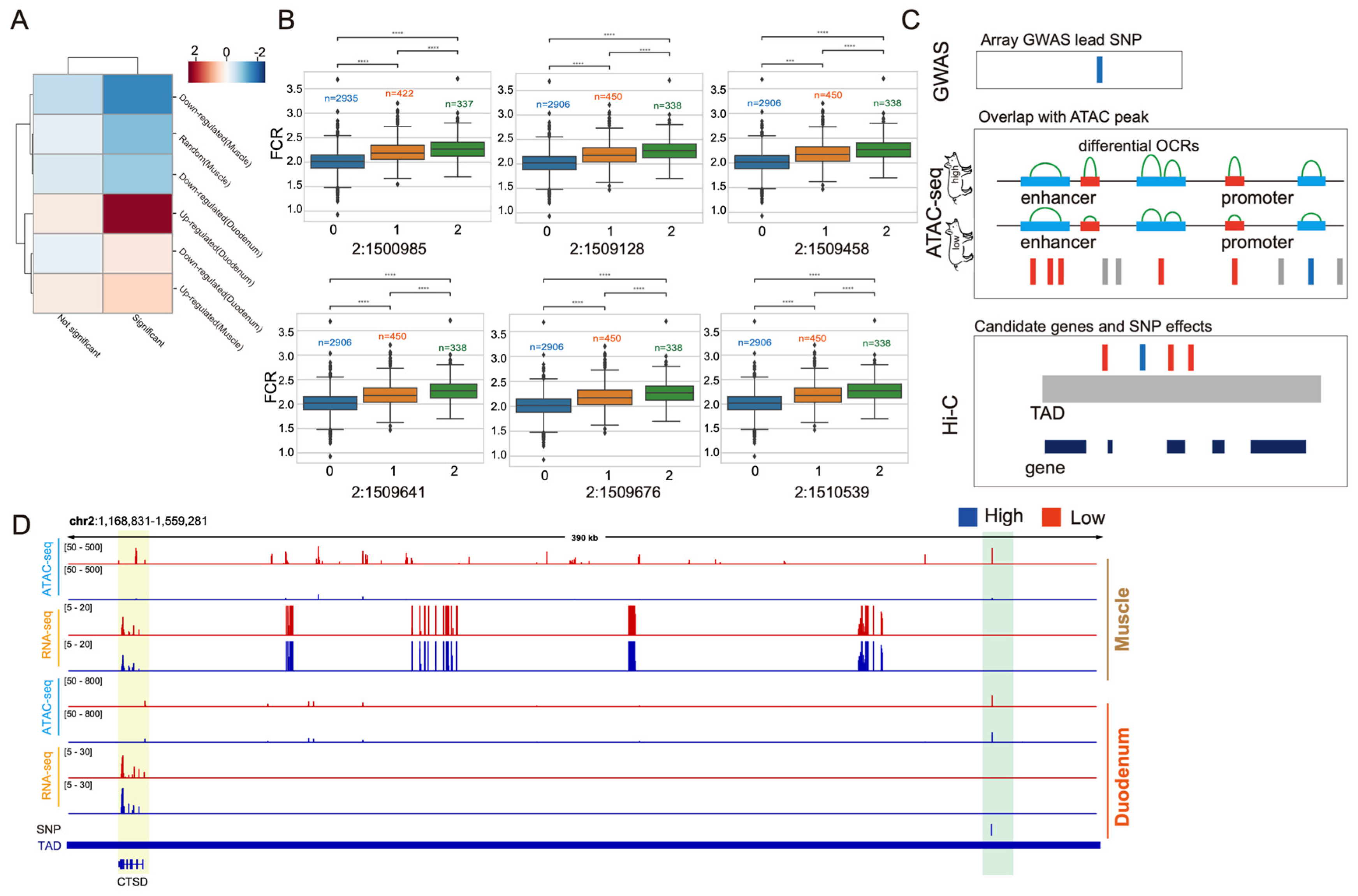
3.4. Integration of Multi-Omics Data for Identification of Key Functional Variants Underlying GWAS Signals
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boliko, M.C. FAO and the Situation of Food Security and Nutrition in the World. J. Nutr. Sci. Vitaminol. 2019, 65, S4–S8. [Google Scholar] [CrossRef] [PubMed]
- Davoudi, P.; Do, D.N.; Colombo, S.M.; Rathgeber, B.; Miar, Y. Application of Genetic, Genomic and Biological Pathways in Improvement of Swine Feed Efficiency. Front. Genet. 2022, 13, 903733. [Google Scholar] [CrossRef] [PubMed]
- Nkrumah, J.; Basarab, J.; Price, M.; Okine, E.; Ammoura, A.; Guercio, S.; Hansen, C.; Li, C.; Benkel, B.; Murdoch, B. Different measures of energetic efficiency and their phenotypic relationships with growth, feed intake, and ultrasound and carcass merit in hybrid cattle. J. Anim. Sci. 2004, 82, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Maltecca, C.; Gray, K.A.; Cassady, J.P. Feed intake, average daily gain, feed efficiency, and real-time ultrasound traits in Duroc pigs: I. Genetic parameter estimation and accuracy of genomic prediction. J. Anim. Sci. 2014, 92, 2377–2386. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Li, T.; Wang, W.; Gao, H.; Bai, Y.; Zhang, S.; Zang, J.; Li, D.; Wang, J. Metabolic characteristics and nutrient utilization in high-feed-efficiency pigs selected using different feed conversion ratio models. Sci. China Life Sci. 2019, 62, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.H.; Xu, J.X.; Zhu, S.W.; Cai, X.; Yang, S.F.; Chen, X.L.; Guo, Q. Gene expression profiling analysis reveals weaning-induced cell cycle arrest and apoptosis in the small intestine of pigs. J. Anim. Sci. 2014, 92, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, L.; Li, P.; Li, X.; Zhou, H.; Wang, F.; Li, D.; Yin, Y.; Wu, G. Gene expression is altered in piglet small intestine by weaning and dietary glutamine supplementation. J. Nutr. 2008, 138, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Guerra, A.; Etienne-Mesmin, L.; Livrelli, V.; Denis, S.; Blanquet-Diot, S.; Alric, M. Relevance and challenges in modeling human gastric and small intestinal digestion. Trends Biotechnol. 2012, 30, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Fändriks, L. Roles of the gut in the metabolic syndrome: An overview. J. Intern. Med. 2017, 281, 319–336. [Google Scholar] [CrossRef]
- Monteiro, M.P.; Batterham, R.L. The importance of the gastrointestinal tract in controlling food intake and regulating energy balance. Gastroenterology 2017, 152, 1707–1717.e1702. [Google Scholar] [CrossRef]
- Jing, L.; Hou, Y.; Wu, H.; Miao, Y.; Li, X.; Cao, J.; Michael Brameld, J.; Parr, T.; Zhao, S. Transcriptome analysis of mRNA and miRNA in skeletal muscle indicates an important network for differential Residual Feed Intake in pigs. Sci. Rep. 2015, 5, 11953. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Mei, Q.; Fu, C.; Liao, M.; Liu, Y.; Xu, X.; Li, X.; Zhao, S.; Xiang, T. Genome-wide association and transcriptome studies identify candidate genes and pathways for feed conversion ratio in pigs. BMC Genom. 2021, 22, 294. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Cheng, D.; Chen, S.; Wang, L.; Li, Y.; Ma, X.; Song, X.; Liu, X.; Li, W.; Liang, J.; et al. Genome-wide association analysis of meat quality traits in a porcine Large White x Minzhu intercross population. Int. J. Biol. Sci. 2012, 8, 580–595. [Google Scholar] [CrossRef]
- Horodyska, J.; Hamill, R.M.; Varley, P.F.; Reyer, H.; Wimmers, K. Genome-wide association analysis and functional annotation of positional candidate genes for feed conversion efficiency and growth rate in pigs. PLoS ONE 2017, 12, e0173482. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Yang, M.; Wang, X.; Quan, J.; Zhuang, Z.; Zhou, S.; Li, S.; Xu, Z.; Zheng, E.; Cai, G. Genetic architecture of feeding behavior and feed efficiency in a Duroc pig population. Front. Genet. 2018, 9, 220. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.F.; Lopes, M.S.; Lopes, P.S.; Gasparino, E. A genome-wide association study for feed efficiency-related traits in a crossbred pig population. Animal 2019, 13, 2447–2456. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K. New members of mammalian aquaporins: AQP10–AQP12. Handb. Exp. Pharmacol. 2009, 190, 251–262. [Google Scholar] [CrossRef]
- Sim, W.C.; Lee, W.; Sim, H.; Lee, K.Y.; Jung, S.H.; Choi, Y.J.; Kim, H.Y.; Kang, K.W.; Lee, J.Y.; Choi, Y.J.; et al. Downregulation of PHGDH expression and hepatic serine level contribute to the development of fatty liver disease. Metabolism 2020, 102, 154000. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Begg, D.P.; Steinbrecher, K.A.; Mul, J.D.; Chambers, A.P.; Kohli, R.; Haller, A.; Cohen, M.B.; Woods, S.C.; Seeley, R.J. Effect of guanylate cyclase-C activity on energy and glucose homeostasis. Diabetes 2014, 63, 3798–3804. [Google Scholar] [CrossRef] [PubMed]
- Le Dily, F.; Bau, D.; Pohl, A.; Vicent, G.P.; Serra, F.; Soronellas, D.; Castellano, G.; Wright, R.H.; Ballare, C.; Filion, G.; et al. Distinct structural transitions of chromatin topological domains correlate with coordinated hormone-induced gene regulation. Genes Dev. 2014, 28, 2151–2162. [Google Scholar] [CrossRef]
- Chedea, V.S.; Palade, L.M.; Marin, D.E.; Pelmus, R.S.; Habeanu, M.; Rotar, M.C.; Gras, M.A.; Pistol, G.C.; Taranu, I. Intestinal Absorption and Antioxidant Activity of Grape Pomace Polyphenols. Nutrients 2018, 10, 588. [Google Scholar] [CrossRef]
- Jian, Y.; Kong, L.; Xu, H.; Shi, Y.; Huang, X.; Zhong, W.; Huang, S.; Li, Y.; Shi, D.; Xiao, Y.; et al. Protein phosphatase 1 regulatory inhibitor subunit 14C promotes triple-negative breast cancer progression via sustaining inactive glycogen synthase kinase 3 β. Clin. Transl. Med. 2022, 12, e725. [Google Scholar] [CrossRef]
- Korner, J.; Cline, G.W.; Slifstein, M.; Barba, P.; Rayat, G.R.; Febres, G.; Leibel, R.L.; Maffei, A.; Harris, P.E. A role for foregut tyrosine metabolism in glucose tolerance. Mol. Metab. 2019, 23, 37–50. [Google Scholar] [CrossRef]
- Sun, J.; MacKinnon, R. Structural Basis of Human KCNQ1 Modulation and Gating. Cell 2020, 180, 340–347.e349. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Heard, E. Structural and functional diversity of topologically associating domains. FEBS Lett. 2015, 589, 2877–2884. [Google Scholar] [CrossRef] [PubMed]
- Thelen, A.M.; Zoncu, R. Emerging Roles for the Lysosome in Lipid Metabolism. Trends Cell Biol. 2017, 27, 833–850. [Google Scholar] [CrossRef]
- Yadati, T.; Houben, T.; Bitorina, A.; Oligschlaeger, Y.; Gijbels, M.J.; Mohren, R.; Lutjohann, D.; Khurana, P.; Goyal, S.; Kulkarni, A.; et al. Inhibition of Extracellular Cathepsin D Reduces Hepatic Lipid Accumulation and Leads to Mild Changes in Inflammationin NASH Mice. Front. Immunol. 2021, 12, 675535. [Google Scholar] [CrossRef]
- Mosley, A.L.; Corbett, J.A.; Ozcan, S. Glucose regulation of insulin gene expression requires the recruitment of p300 by the β-cell-specific transcription factor Pdx-1. Mol. Endocrinol. 2004, 18, 2279–2290. [Google Scholar] [CrossRef]
- Fontanesi, L.; Speroni, C.; Buttazzoni, L.; Scotti, E.; Dall’Olio, S.; Nanni Costa, L.; Davoli, R.; Russo, V. The insulin-like growth factor 2 (IGF2) gene intron3-g.3072G>A polymorphism is not the only Sus scrofa chromosome 2p mutation affecting meat production and carcass traits in pigs: Evidence from the effects of a cathepsin D (CTSD) gene polymorphism. J. Anim. Sci. 2010, 88, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Wijtten, P.J.; van der Meulen, J.; Verstegen, M.W. Intestinal barrier function and absorption in pigs after weaning: A review. Br. J. Nutr. 2011, 105, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Uni, Z.; Smirnov, A.; Sklan, D. Pre- and posthatch development of goblet cells in the broiler small intestine: Effect of delayed access to feed. Poult. Sci. 2003, 82, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Saif, L.J. Goblet cell depletion in small intestinal villous and crypt epithelium of conventional nursing and weaned pigs infected with porcine epidemic diarrhea virus. Res. Vet. Sci. 2017, 110, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Masaratana, P.; Latunde-Dada, G.O.; Arno, M.; Simpson, R.J.; McKie, A.T. Duodenal reductase activity and spleen iron stores are reduced and erythropoiesis is abnormal in Dcytb knockout mice exposed to hypoxic conditions. J. Nutr. 2012, 142, 1929–1934. [Google Scholar] [CrossRef] [PubMed]
- Busslinger, G.A.; Weusten, B.L.; Bogte, A.; Begthel, H.; Brosens, L.A.; Clevers, H. Human gastrointestinal epithelia of the esophagus, stomach, and duodenum resolved at single-cell resolution. Cell Rep. 2021, 34, 108819. [Google Scholar] [CrossRef]
- Wang, Y.; Song, W.; Wang, J.; Wang, T.; Xiong, X.; Qi, Z.; Fu, W.; Yang, X.; Chen, Y.G. Single-cell transcriptome analysis reveals differential nutrient absorption functions in human intestine. J. Exp. Med. 2020, 217, e20191130. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, Y.; Sun, J.; Ma, G.; Dai, X.; Meng, Y.; Fu, C.; Zhang, Y.; Zhao, Q.; Li, J.; Zhang, S.; et al. Integrating Multi-Omics Data to Identify Key Functional Variants Affecting Feed Efficiency in Large White Boars. Genes 2024, 15, 980. https://doi.org/10.3390/genes15080980
Xiang Y, Sun J, Ma G, Dai X, Meng Y, Fu C, Zhang Y, Zhao Q, Li J, Zhang S, et al. Integrating Multi-Omics Data to Identify Key Functional Variants Affecting Feed Efficiency in Large White Boars. Genes. 2024; 15(8):980. https://doi.org/10.3390/genes15080980
Chicago/Turabian StyleXiang, Yue, Jiahui Sun, Guojian Ma, Xueting Dai, Yuan Meng, Chong Fu, Yan Zhang, Qiulin Zhao, Jingjin Li, Saixian Zhang, and et al. 2024. "Integrating Multi-Omics Data to Identify Key Functional Variants Affecting Feed Efficiency in Large White Boars" Genes 15, no. 8: 980. https://doi.org/10.3390/genes15080980