
Impact of NSD1 Alternative Transcripts in Actin Filament Formation and Cellular Division Pathways in Fibroblasts
 , , , , , and
, , , , , and
Abstract
:1. Introduction
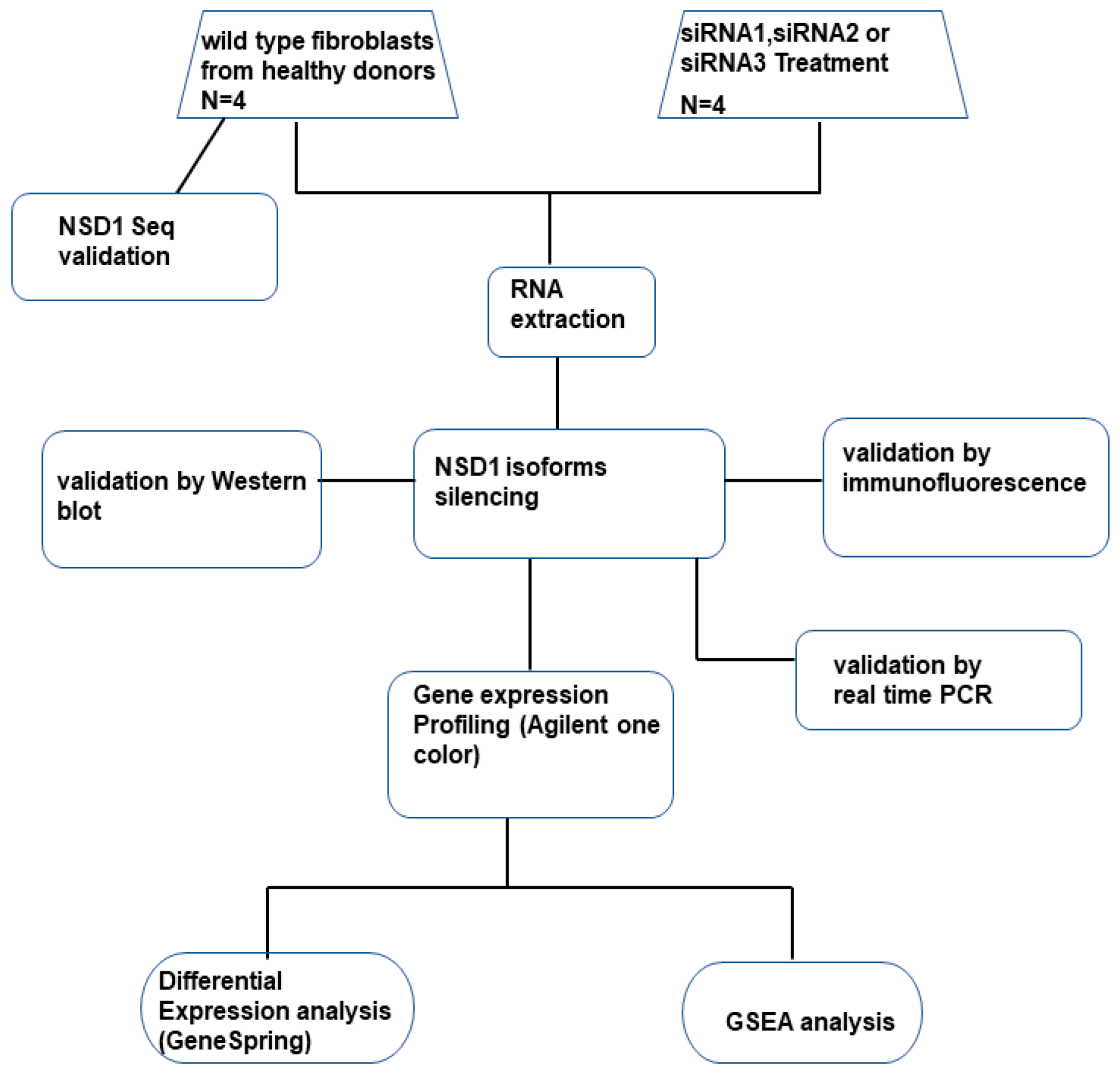
2. Materials and Methods
2.1. Patients
2.2. Cell Culture
2.3. Analysis of NSD1 Sequence
2.4. siRNA Transfection
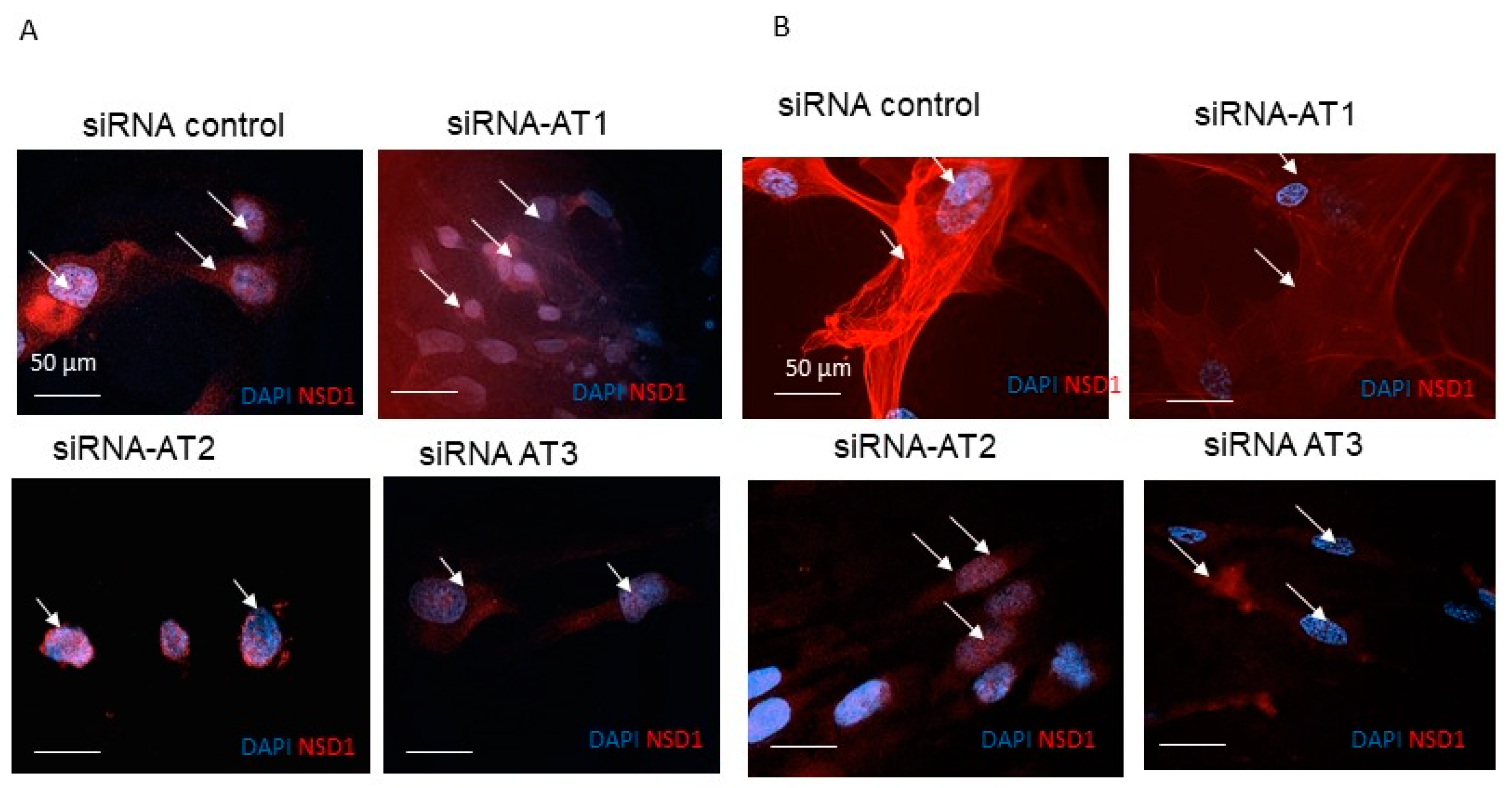
2.5. Indirect Immunofluorescence
2.6. Gene Expression Profiling
2.7. Quantitative Real-Time RT–PCR
2.8. Western Blotting
2.9. Bioinformatic Analysis
3. Results
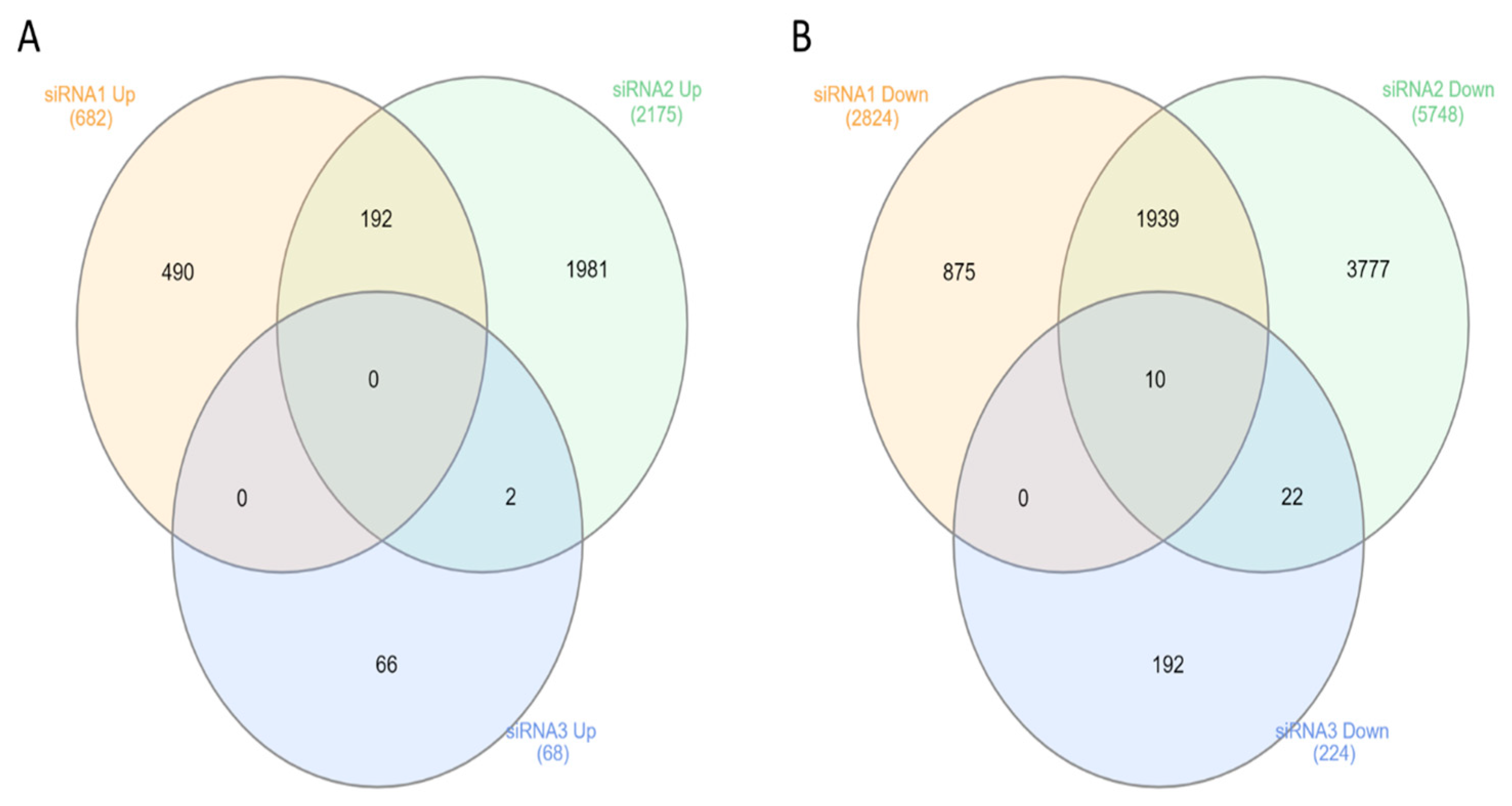
3.1. Effect of Anti-NSD1 siRNAs on NSD1 Isoform Expression
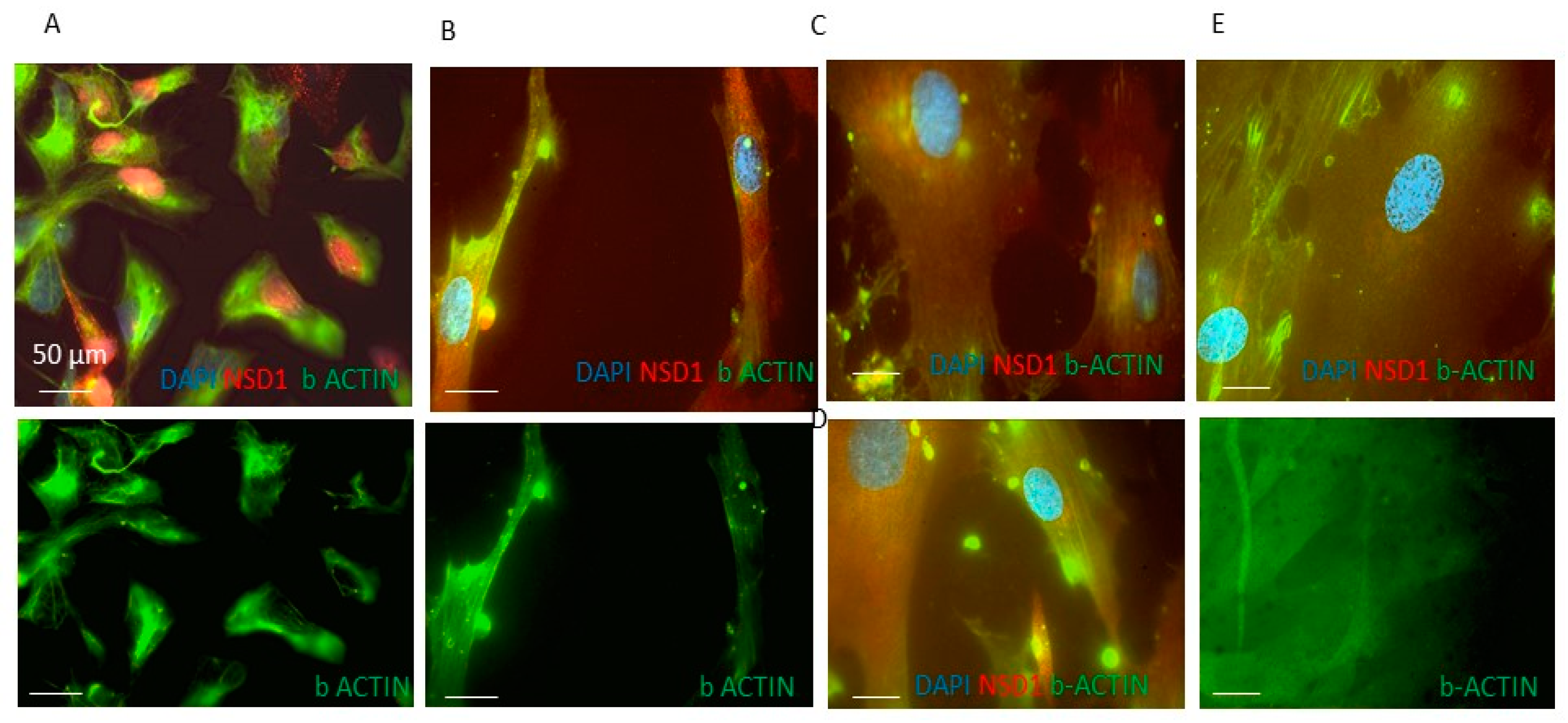
3.2. NSD1 Protein Isoform AT3 Exhibits Distinct Subcellular Localization with Respect to the Long Canonical Isoform in Fibroblasts
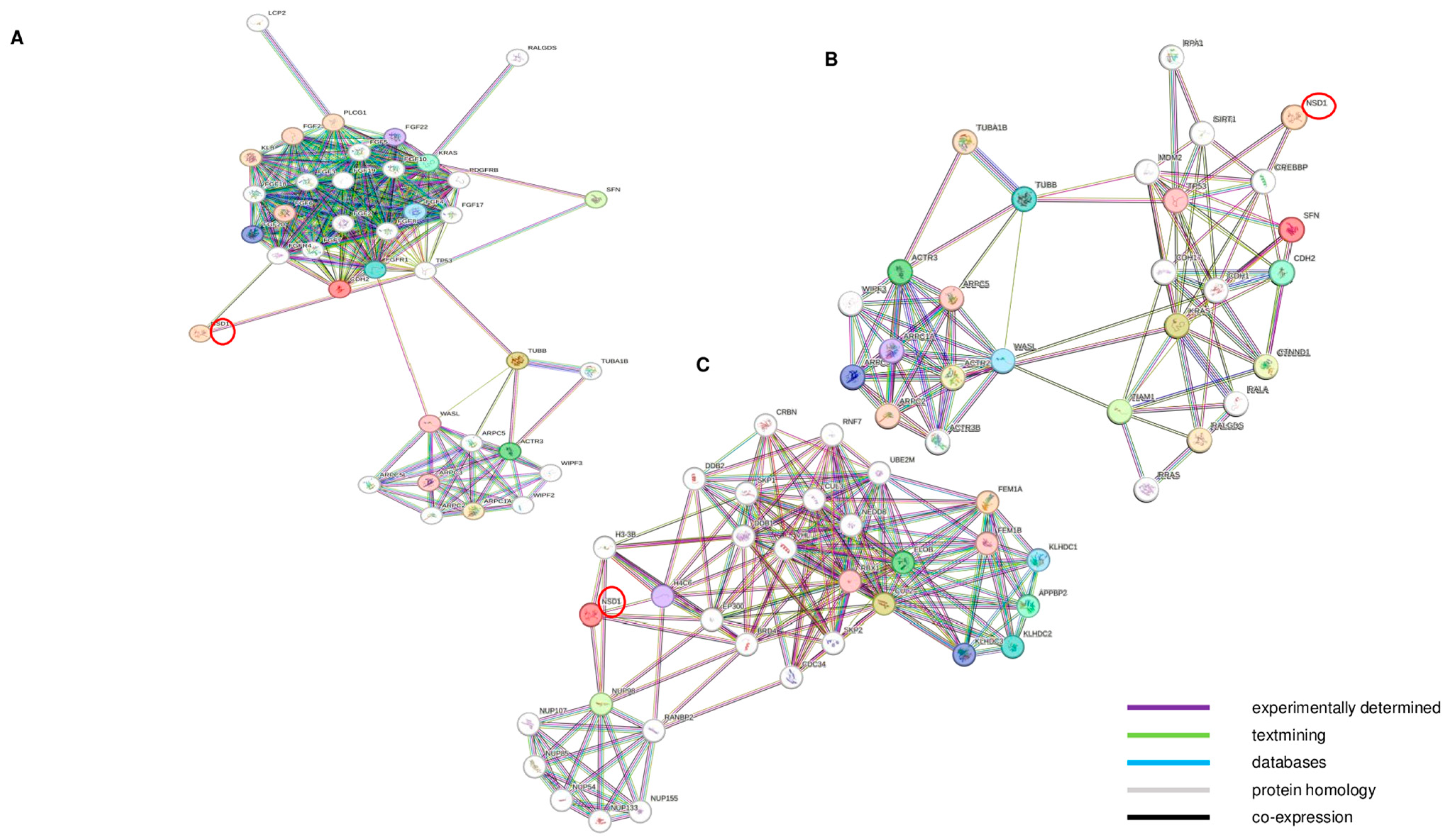
3.3. NSD1 AT2 and NSD1 AT3 Isoforms May Mediate Transcriptomic Effects on BCL2, KRAS and p53 Signaling Pathways
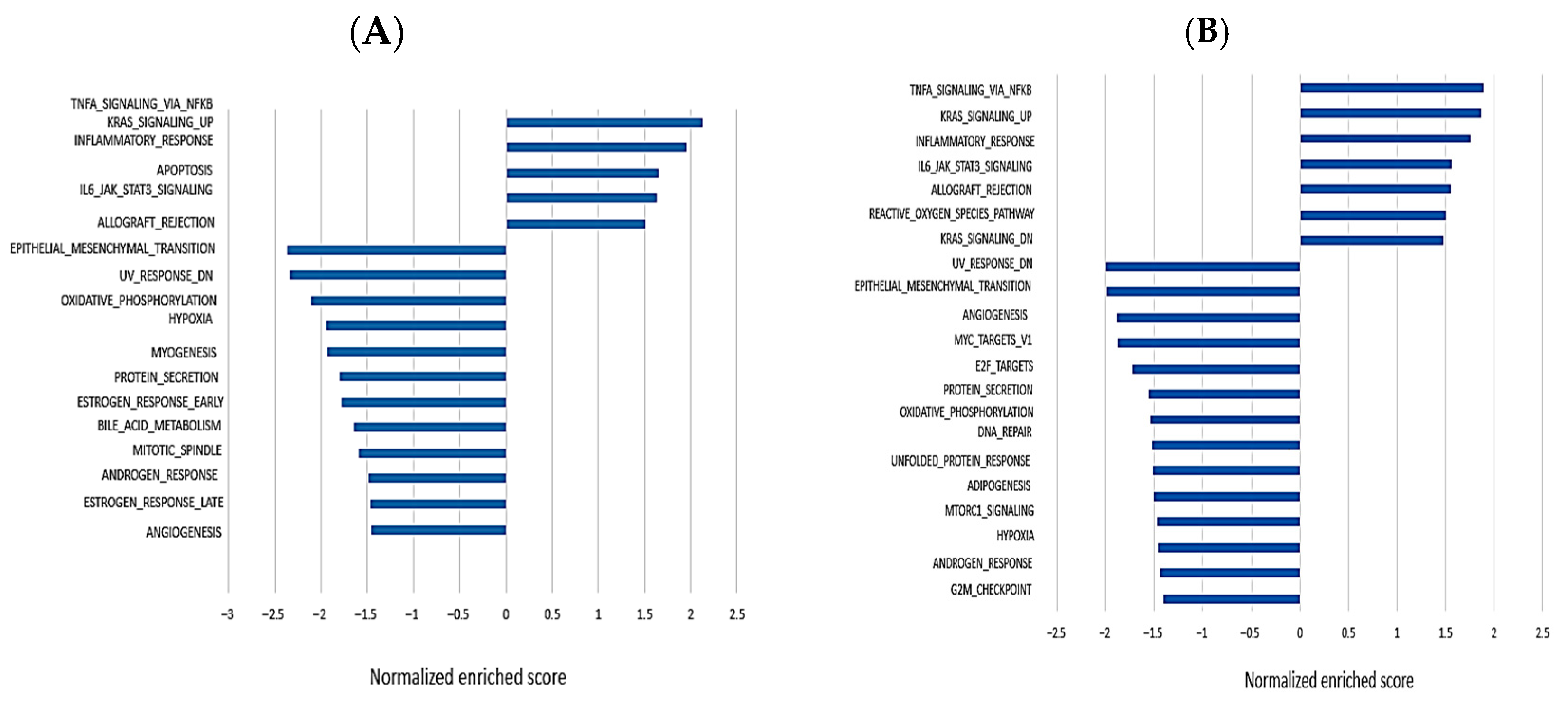
3.4. Gene Set Enrichment Analysis to Perform an Unbiased Biological Interpretation of the Data
3.5. NSD1 Is Involved in the Cytoskeleton Actin Stress Fiber (SFs) Organization Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lane, C.; Milne, E.; Freeth, M.J. Characteristics of Autism Spectrum Disorder in Sotos Syndrome. Autism. Dev. Disord. 2017, 47, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Conteduca, G.; Grasso, M.; Cecconi, M.; Lantieri, F.; Baldo, C.; Arado, A.; Andraghetti, L.; Malacarne, M.; Milani, D.; et al. Molecular Analysis and Reclassification of NSD1 Gene Variants in a Cohort of Patients with Clinical Suspicion of Sotos Syndrome. Genes 2022, 14, 295. [Google Scholar] [CrossRef]
- Sotos, J.F.; Dodge, P.R.; Muirhead, D.; Crawford, J.D.; Talbot, N.B. Cerebral gigantism in childhood: A syndrome of excessively rapid growth with acromegalic features and a nonprogressive neurologic disorder. N. Engl. J. Med. 1964, 271, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Rayasam, G.V.; Wendling, O.; Angrand, P.O.; Mark, M.; Niederreither, K.; Song, L.; Lerouge, T.; Hager, G.L.; Chambon, P.; Losson, R. NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO J. 2003, 22, 3153–3163. [Google Scholar] [CrossRef] [PubMed]
- Hamagami, N.; Wu, D.Y.; Clemens, A.W.; Nettles, S.A.; Li, A.; Gabel, H.W. NSD1 deposits histone H3 lysine 36 dimethylation to pattern non-CG DNA methylation in neurons. Mol. Cell. 2023, 83, 1412–1428.e7. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhao, C.; Song, Q.; Xu, L.; Zhang, B.; Hu, G.; Kong, X.; Li, S.; Li, X.; Shen, Y.; et al. Histone methylation mediated by NSD1 is required for the establishment and maintenance of neuronal identities. Cell Rep. 2023, 42, 113496. [Google Scholar] [CrossRef]
- Lucio-Eterovic, A.K.; Singh, M.M.; Gardner, J.E.; Veerappan, C.S.; Rice, J.C.; Carpenter, P.B. Role for the nuclear receptor-binding SET domain protein 1 (NSD1) methyltransferase in coordinating lysine 36 methylation at histone 3 with RNA polymerase II function. Proc. Natl. Acad. Sci. USA 2010, 107, 16952–16957. [Google Scholar] [CrossRef]
- Choufani, S.; Cytrynbaum, C.; Chung, B.H.; Turinsky, A.L.; Grafodatskaya, D.; Chen, Y.A.; Cohen, A.S.; Dupuis, L.; Butcher, D.T.; Siu, M.T.; et al. NSD1 mutations generate a genome-wide DNA methylation signature. Nat. Commun. 2015, 6, 10207. [Google Scholar] [CrossRef]
- Chen, H.; Hu, B.; Horth, C.; Bareke, E.; Rosenbaum, P.; Kwon, S.Y.; Sirois, J.; Weinberg, D.N.; Robison, F.M.; Garcia, B.A.; et al. H3K36 dimethylation shapes the epigenetic interaction landscape by directing repressive chromatin modifications in embryonic stem cells. Genome Res. 2022, 32, 825–837. [Google Scholar] [CrossRef]
- Foster, A.; Zachariou, A.; Loveday, C.; Ashraf, T.; Blair, E.; Clayton-Smith, J. The phenotype of Sotos syndrome in adulthood: A review of 44 individuals. Am. J. Med. Genet. 2019, 181, 502–508. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Conteduca, B.; Testa, C.; Baldo, A.; Arado, M.; Malacarne, G.; Candiano, A.; Garbarino, D.A.; Coviello, C.; Cantoni, C. NSD1 Identification of alternative transcripts of NSD1 gene in Sotos Syndrome patients and healthy subjects. Gene 2023, 851, 146970. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; vom Baur, E.; Garnier, J.M.; Lerouge, T.; Vonesch, J.L.; Lutz, Y.; Chambon, P.; Losson, R. Two distinct nuclear receptor interaction domains in NSD1, a novel SET protein that exhibits characteristics of both corepressors and coactivators. EMBO J. 1998, 15, 3398–3412. [Google Scholar] [CrossRef] [PubMed]
- Neverov, A.D.; Artamonova, I.I.; Nurtdinov, R.N.; Frishman, D.; Gelfand, M.S.; Mironov, A.A. Alternative splicing and protein function. BMC Bioinform. 2005, 6, 266. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef]
- Rhine, C.L.; Cygan, K.J.; Soemedi, R.; Maguire, S.; Murray, M.F.; Monaghan, S.F.; Fairbrother, W.G. Hereditary cancer genes are highly susceptible to splicing mutations. PLoS Genet. 2018, 14, e1007231. [Google Scholar] [CrossRef]
- Kurotaki, N.; Imaizumi, K.; Harada, N.; Masuno, M.; Kondoh, T.; Nagai, T.; Ohashi, H.; Naritomi, K.; Tsukahara, M.; Makita, Y.; et al. Molecular characterization of NSD1, a human homologue of the mouse Nsd1 gene. Gene 2021, 279, 197–204. [Google Scholar] [CrossRef]
- Conteduca, G.; Cangelosi, D.; Coco, S.; Malacarne, M.; Baldo, C.; Arado, A.; Pinto, R.; Testa, B.; Coviello, D.A. NSD1 Mutations in Sotos Syndrome Induce Differential Expression of Long Noncoding RNAs, miR646 and Genes Controlling the G2/M Checkpoint. Life 2022, 12, 988. [Google Scholar] [CrossRef]
- Gaur, R.K. RNA interference: A potential therapeutic tool for silencing splice isoforms linked to human diseases. Biotechniques 2006, 40, S15–S22. [Google Scholar] [CrossRef]
- Kisielow, M.; Kleiner, S.; Nagasawa, M.; Faisal, A.; Nagamine, Y. Isoform-specific knockdown and expression of adaptor protein ShcA using small interfering RNA. Biochem. J. 2002, 363 Pt 1, 1–5. [Google Scholar] [CrossRef]
- Livak, J.K.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)). Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8—A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412–D416. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Farhangdoost, N.; Horth, C.; Hu, B.; Bareke, E.; Chen, X.; Li, Y.; Coradin, M.; Garcia, B.A.; Lu, C.; Majewski, J. Chromatin dysregulation associated with NSD1 mutation in head and neck squamous cell carcinoma. Cell Rep. 2021, 34, 108769. [Google Scholar] [CrossRef]
- Yang, C.; Wang, K.; Liang, Q.; Tian, T.T.; Zhong, Z. Role of NSD1 as potential therapeutic target in tumor. Pharmacol. Res. 2021, 173, 105888. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yu, H.; Xu, J.; Zhou, F.; Tang, S.; Feng, X.; Luo, Q.; Zhang, B.; Wu, X.; Jin, R.; et al. Refractory pediatric acute myeloid leukemia expressing NUP98-NSD1 fusion gene responsive to chemotherapy combined with venetoclax and decitabine. Pediatr. Blood Cancer 2023, 70, e30021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wen, H.; Shi, X. Lysine Methylation: Beyond Histones. Acta Biochim. Biophys. Sin. 2012, 44, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, S.M.; Wilkinson, A.W.; Elias, J.E.; Gozani, O. A PWWP Domain of Histone-Lysine N-Methyltransferase NSD2 Binds to Dimethylated Lys-36 of Histone H3 and Regulates NSD2 Function at Chromatin. J. Biol. Chem. 2016, 291, 8465–8474. [Google Scholar] [CrossRef]
- Su, X.; Zhang, J.; Mouawad, R.; Compérat, E.; Rouprêt, M.; Allanic, F.; Parra, J.; Bitker, M.O.; Thompson, E.J. NSD1 Inactiva tion and SETD2 Mutation Drive a Convergence toward Loss of Function of H3K36 Writers in Clear Cell Renal Cell Carcinomas. Cancer Res. 2017, 77, 4835–4845. [Google Scholar] [CrossRef]
- Mohammad, F.; Helin, K. Oncohistones: Drivers of pediatric cancers. Genes Dev. 2017, 31, 2313–2324. [Google Scholar] [CrossRef]
- Schuhmacher, M.; Kusevic, D.; Kudithipudi, S.; Jeltsch, A. Kinetic Analysis of the Inhibition of the NSD1, NSD2 and SETD2 Protein Lysine Methyltransferases by a K36M Oncohistone Peptide. Chem. Select. 2017, 2, 9532–9536. [Google Scholar] [CrossRef]
- Oishi, S.; Zalucki, O.; Vega, M.S.; Harkins, D.; Harvey, T.J.; Kasherman, M.; Davila, R.A.; Hale, L.; White, M.; Piltz, S.; et al. Investigating cortical features of Sotos syndrome using mice heterozygous for Nsd1. Genes Brain Behav. 2020, 19, e12637. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Izreig, S.; Yarbrough, W.G.; Issaeva, N. NSD1 Mutations by HPV Status in Head and Neck Cancer: Differences in Survival and Response to DNA-Damaging Agents. Cancers Head Neck 2019, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Bambury, R.M.; Jordan, E.; Zabor, E.C.; Bouvier, N.; Ahmadie, H.A.; Boyd, M.E.; Mullane, S.E.; Cha, E.K.; Ostrovnaya, I.; Hyman, D.M.; et al. Association of Somatic Mutations in DNA Damage Repair (DDR) Genes with Efficacy of Platinum-Based Chemotherapy in Advanced Urothelial Carcinoma. J. Clin. Oncol. 2015, 33, 4532. [Google Scholar] [CrossRef]
- Ceccaldi, R.; O’Connor, K.W.; Mouw, K.W.; Li, A.Y.; Matulonis, U.A.; D’Andrea, A.D.; Konstantinopoulos, P.A. Unique Subset of Epithelial Ovarian Cancers with Platinum Sensitivity and PARP Inhibitor Resistance. Cancer Res. 2015, 75, 628. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, F.; Chen, Q.; Wan, C.; Xiong, J.; Xu, J. CRISPR/Cas9-mediated knockout of NSD1 suppresses the hepatocellular carcinoma development via the NSD1/H3/Wnt10b signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 467. [Google Scholar] [CrossRef]
- Conteduca, G.; Baldo, C.; Arado, A.; Traverso, M.; Testa, B.; Malacarne, M.; Coviello, D.; Zara, F.; Baldassari, S. Generation of induced pluripotent stem cell lines from a patient with Sotos syndrome carrying 5q35 microdeletion. Stem Cell Res. 2023, 66, 103007. [Google Scholar] [CrossRef]
- Conteduca, G.; Baldo, C.; Arado, A.; da Silva, J.S.M.; Testa, B.; Baldassari, S.; Zara, F.; Filaci, G.; Coviello, D.; Malacarne, M. Generation of IGGi003-A induced pluripotent stem cell line from a patient with Sotos Syndrome carrying c.1633delA NSD1 variant in exon 5. Stem Cell Res. 2024, 76, 103324. [Google Scholar] [CrossRef]
- Spurr, L.; Li, M.; Alomran, N.; Zhang, Q.; Restrepo, P.; Movassagh, M.; Trenkov, C.; Tunnessen, N.; Apanasovich, T.; Crandall, K.A.; et al. Systematic pan-cancer analysis of somatic allele frequency. Sci. Rep. 2018, 8, 7735. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Nash, K.T.; Welch, D.R. The KISS1 metastasis suppressor: Mechanistic insights and clinical utility. Front. Biosci. 2006, 11, 647–659. [Google Scholar] [CrossRef]
- Ji, K.; Ye, L.; Mason, M.D.; Jiang, W.J. The Kiss-1/Kiss-1R complex as a negative regulator of cell motility and cancer metastasis (Review). Int. J. Mol. Med. 2013, 32, 747–754. [Google Scholar] [CrossRef]
- Ji, K.; Ye, L.; Ruge, F.; Hargest, R.; Mason, M.D.; Jiang, W.G. Implication of metastasis suppressor gene, Kiss-1 and its receptor Kiss-1R in colorectal cancer. BMC Cancer 2014, 14, 723. [Google Scholar] [CrossRef]
- Chen, Y.; Li, X.; Xu, J.; Xiao, H.; Tang, C.; Liang, W.; Zhu, X.; Fang, Y.; Wang, H.; Shi, J. Knockdown of nuclear receptor binding SET domain-containing protein 1 (NSD1) inhibits proliferation and facilitates apoptosis in paclitaxel-resistant breast cancer cells via inactivating the Wnt/β-catenin signaling pathway. J. Bioengineered. 2022, 13, 3526–3536. [Google Scholar] [CrossRef] [PubMed]
- Koren, I.; Timms, R.T.; Kula, T.; Xu, Q.; Li, M.Z.; Elledge, S.J. The Eukaryotic Proteome Is Shaped by E3 Ubiquitin Ligases Targeting C-Terminal Degrons. Cell 2018, 173, 1622–1635.e14. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, H.; Fujioka, H.; Togashi, K.; Thompson, J.; Yates, J.R.; Gleeson, J.G.; Emoto, K.D. CLK1 phosphorylates the microtubule-associated protein MAP7D1 to promote axon elongation in cortical neurons. Dev. Neurobiol. 2017, 77, 493–510. [Google Scholar] [CrossRef]
- Vijai, M.; Baba, M.; Ramalingam, S.; Thiyagaraj, A. DCLK1 and its interaction partners: An effective therapeutic target for colorectal cancer. Oncol. Lett. 2021, 22, 850. [Google Scholar] [CrossRef]
- Koizumi, H.; Tanaka, T.; Gleeson, J.G. Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron 2005, 49, 55–66. [Google Scholar] [CrossRef]
- Verhey, K.J.; Gaertig, J. The tubulin code. Cell Cycle 2007, 6, 2152–2160. [Google Scholar] [CrossRef]
- Barisic, M.; Silva e Sousa, R.; Tripathy, S.K.; Magiera, M.M.; Zaytsev, A.V.; Pereira, A.L.; Janke, C.; Grishchuk, E.L.; Maiato, H. Mitosis. Microtubule detyrosination guides chromosomes during mitosis. Science 2015, 348, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Janke, C. The tubulin code: Molecular components, readout mechanisms, and functions. J. Cell Biol. 2014, 206, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Park, I.Y.; Powell, R.T.; Tripathi, D.N.; Dere, R.; Ho, T.H.; Blasius, T.L.; Chiang, Y.C.; Davis, I.J.; Fahey, C.C.; Hacker, K.E.; et al. Dual Chromatin and Cytoskeletal Remodeling by SETD2. Cell 2016, 166, 950–962. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probe Set ID | Gene Symbol | Gene Name | Seq. Name | Log2 Fold Change | p-Value | p-Value Adjusted |
|---|---|---|---|---|---|---|
| A_33_P3389286 | SFN | stratifin | NM_006142 | 5.2 | 1.61 × 10−1 | 0.002 |
| A_33_P3321205 | BEGAIN | brain-enriched guanylate kinase-associated | NM_001159531 | 1 | 0.002 | 0.011 |
| A_33_P3363898 | TUBG1 | tubulin, γ 1 | NM_001070 | −0.29 | 0.011 | 0.03 |
| A_32_P377880 | GDNF | glial cell derived neurotrophic factor | NM_001190468 | −0.4 | 0.009 | 0.025 |
| A_23_P80694 | ACTR8 | ARP8 actin-related protein 8 homolog | NM_022899 | −0.5 | 0.003 | 0.014 |
| A_23_P377376 | ACTR2 | ARP2 actin-related protein 2 homolog | NM_001005386 | −0.5 | 0.003 | 0.013 |
| A_33_P3330283 | SP1 | Sp1 transcription factor | NM_138473 | −0.6 | 0.022 | 0.049 |
| A_24_P726336 | PHACTR2 | phosphatase and actin regulator 2 | NM_001100164 | −0.6 | 0.018 | 0.043 |
| A_33_P3363674 | NFYC | Nuclear transcription factor Y | NM_001142590 | −0.7 | 0.0019 | 0.010 |
| A_33_P3260016 | NEDD1 | neural precursor cell expressed, developmentally down-regulated 1 | NM_152905 | −0.7 | 0.003 | 0.014 |
| A_24_P251969 | FGF1 | fibroblast growth factor 1 | NM_000800 | −0.8 | 1.13 × 10−2 | 0.029 |
| A_23_P153256 | FGF3 | fibroblast growth factor 3 | NM_005247 | −0.8 | 8.64 × 10−6 | 0.035 |
| A_23_P123193 | ACTR3B | ARP3 actin-related protein 3 homolog B | NM_020445 | −0.9 | 2.3 × 10−2 | 0.002 |
| A_23_P162596 | ACTR6 | ARP6 actin-related protein 6 homolog | NM_022496 | −1 | 0.006 | 0.019 |
| A_23_P311201 | ACTR3 | ARP3 actin-related protein 3 homolog | NM_005721 | −1.1 | 6.2 × 10−2 | 0.031 |
| A_23_P208961 | KRAS | Kirsten rat sarcoma viral oncogene homolog (KRAS) | NM_004985 | −1.1 | 8.00 × 10−6 | 0.039 |
| A_33_P3363799 | NCAM1 | neural cell adhesion molecule 1 | NM_001242607 | −3.1 | 0.011 | 0.030 |
| A_22_P00001924 | DDB1 | damage-specific DNA binding protein 1 | NM_001923 | −3.9 | 7.04 × 10−2 | 0.029 |
| Probe Set ID | Gene Symbol | Gene Name | Seq. Name | Log2 Fold Change | p-Value | p-Value Adjusted |
|---|---|---|---|---|---|---|
| A_23_P77493 | TUBB3 | tubulin, β 3 class III | NM_006086 | 1 | 0.002 | 0.006 |
| A_23_P102122 | ARPC2 | actin-related protein 2/3 complex, subunit 2, 34 kDa | NM_152862 | −0.2 | 0.007 | 0.010 |
| A_23_P377376 | ACTR2 | ARP2 actin-related protein 2 | NM_001005386 | −0.4 | 7.9 × 10−3 | 0.004 |
| A_24_P167473 | ARPC3 | actin-related protein 2/3 complex, subunit 3, 21 kDa | NM_001278556 | −0.5 | 0.002 | 0.006 |
| A_33_P3416946 | ACTR10 | actin-related protein 10 homolog | NM_018477 | −0.7 | 0.013 | 0.014 |
| A_24_P72479 | ARPC1A | actin-related protein 2/3 complex, subunit 1A, 41 kDa | NM_006409 | −0.8 | 0.005 | 0.009 |
| A_23_P19291 | TUBB2A | tubulin, β 2A class IIa | NM_001069 | −0.8 | 0.002 | 0.006 |
| A_33_P3338909 | ARPC5 | actin-related protein 2/3 complex, subunit 5, 16 kDa | NM_001270439 | −0.9 | 9.12 × 10−2 | 0.004 |
| A_23_P60283 | PHACTR2 | phosphatase and actin regulator 2 | NM_014721 | −1 | 0.001 | 0.005 |
| A_21_P0014389 | ACTR3 | ARP3 actin-related protein 3 homolog | NM_005721 | −1 | 1.96 × 10−3 | 0.003 |
| A_23_P162596 | ACTR6 | ARP6 actin-related protein 6 homolog | NM_022496 | −1 | 5.24 × 10−2 | 0.002 |
| A_23_P123193 | ACTR3B | ARP3 actin-related protein 3 homolog B | NM_005247 | −1.1 | 0.002 | 0.007 |
| Probe Set ID | Gene Symbol | Gene Name | Seq. Name | Log2 Fold Change | p-Value | p-Value Adjusted |
|---|---|---|---|---|---|---|
| A_23_P344555 | NEDD9 | neural precursor cell expressed, developmentally down-regulated 9 | NM_006403 | −0.05 | 1.13 × 10−2 | 0.0013 |
| A_33_P3270009 | NCOA1 | nuclear receptor coactivator 1 | NM_147223 | −0.4 | 8.13 × 10−1 | 0.01 |
| A_32_P75902 | MEIOB | meiosis specific with OB domains | NM_152764 | −0.6 | 8.08 × 10−8 | 0.01 |
| A_23_P76774 | GSC | goosecoid homeobox | NM_173849 | −1.1 | 1.92 × 10−2 | 0.002 |
| A_33_P3302389 | FEM1A | fem−1 homolog a | NM_018708 | −1.3 | 2.22 × 10−10 | 1.74 × 10−6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conteduca, G.; Cangelosi, D.; Baldo, C.; Arado, A.; Testa, B.; Wagner, R.T.; Robertson, K.D.; Dequiedt, F.; Fitzsimmons, L.; Malacarne, M.; et al. Impact of NSD1 Alternative Transcripts in Actin Filament Formation and Cellular Division Pathways in Fibroblasts. Genes 2024, 15, 1117. https://doi.org/10.3390/genes15091117
Conteduca G, Cangelosi D, Baldo C, Arado A, Testa B, Wagner RT, Robertson KD, Dequiedt F, Fitzsimmons L, Malacarne M, et al. Impact of NSD1 Alternative Transcripts in Actin Filament Formation and Cellular Division Pathways in Fibroblasts. Genes. 2024; 15(9):1117. https://doi.org/10.3390/genes15091117
Chicago/Turabian StyleConteduca, Giuseppina, Davide Cangelosi, Chiara Baldo, Alessia Arado, Barbara Testa, Ryan T. Wagner, Keith D. Robertson, Franck Dequiedt, Lane Fitzsimmons, Michela Malacarne, and et al. 2024. "Impact of NSD1 Alternative Transcripts in Actin Filament Formation and Cellular Division Pathways in Fibroblasts" Genes 15, no. 9: 1117. https://doi.org/10.3390/genes15091117