
A 10-Year Review on Advancements in Identifying and Treating Intellectual Disability Caused by Genetic Variations

Abstract
:1. Introduction
2. Identification
3. Physiological Basis
4. Genetic Etiology of ID
4.1. Autosomal Dominant ID
4.2. Autosomal Recessive ID
4.3. XLID
5. Treatment
5.1. General Treatment
5.2. Genetic Therapy
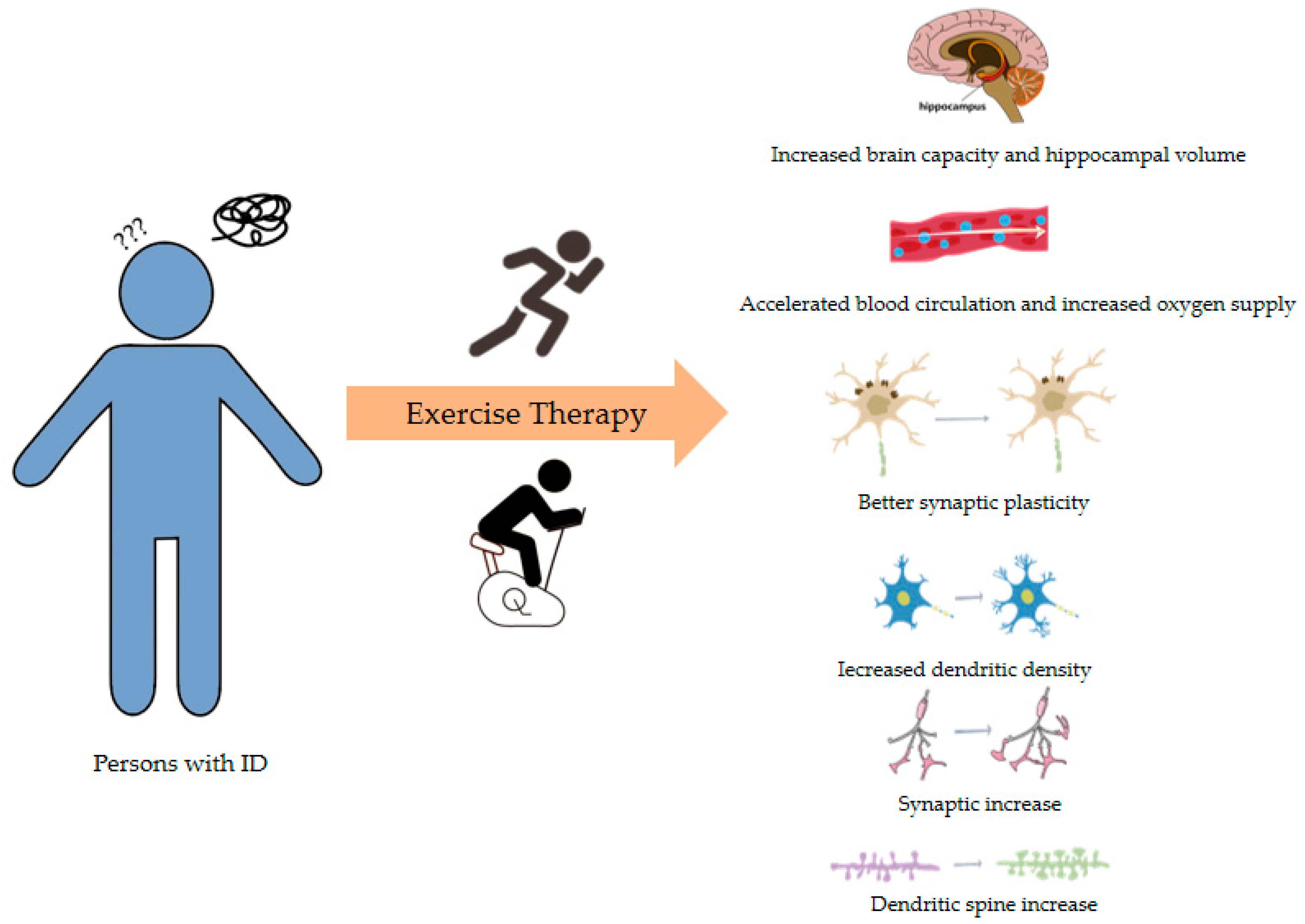
5.3. Exercise Therapy
6. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Schwartz, C.E.; Louie, R.J.; Toutain, A.; Skinner, C.; Friez, M.J.; Stevenson, R.E. X-Linked intellectual disability update 2022. Am. J. Med. Genet. A 2023, 191, 144–159. [Google Scholar] [CrossRef]
- Belkady, B.; Elkhattabi, L.; Elkarhat, Z.; Zarouf, L.; Razoki, L.; Aboulfaraj, J.; Nassereddine, S.; Cadi, R.; Rouba, H.; Barakat, A. Chromosomal Abnormalities in Patients with Intellectual Disability: A 21-Year Retrospective Study. Hum. Hered. 2018, 83, 274–282. [Google Scholar] [CrossRef]
- Almeida, L.O.; Neto, M.P.; Sousa, L.O.; Tannous, M.A.; Curti, C.; Leopoldino, A.M. SET oncoprotein accumulation regulates transcription through DNA demethylation and histone hypoacetylation. Oncotarget 2017, 8, 26802. [Google Scholar] [CrossRef]
- Jansen, S.; Vissers, L.E.; de Vries, B.B. The Genetics of Intellectual Disability. Brain Sci. 2023, 13, 231. [Google Scholar] [CrossRef]
- Nelson, D.L. Mental retardation and intellectual disability. In Vogel and Motulsky’s Human Genetics; Springer: Berlin/Heidelberg, Germany, 2010; pp. 663–680. [Google Scholar]
- Moeschler, J.B.; Shevell, M.; Genetics, C.O.; Moeschler, J.B.; Shevell, M.; Saul, R.A.; Chen, E.; Freedenberg, D.L.; Hamid, R.; Jones, M.C. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics 2014, 134, e903–e918. [Google Scholar] [CrossRef]
- Katz, G.; Lazcano-Ponce, E. Intellectual disability: Definition, etiological factors, classification, diagnosis, treatment and prognosis. Salud Pública México 2008, 50, 132–141. [Google Scholar] [CrossRef]
- Xiao, T.; Zhou, W. The third generation sequencing: The advanced approach to genetic diseases. Transl. Pediatr. 2020, 9, 163. [Google Scholar] [CrossRef]
- McCombie, W.R.; McPherson, J.D.; Mardis, E.R. Next-generation sequencing technologies. Cold Spring Harb. Perspect. Med. 2019, 9, a36798. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, P.; Wang, D.; Gu, W.; Wang, K. Interrogating the “unsequenceable” genomic trinucleotide repeat disorders by long-read sequencing. Genome Med. 2017, 9, 1–16. [Google Scholar] [CrossRef]
- Mastrorosa, F.K.; Miller, D.E.; Eichler, E.E. Applications of long-read sequencing to Mendelian genetics. Genome Med. 2023, 15, 42. [Google Scholar] [CrossRef]
- Zhao, X.; Collins, R.L.; Lee, W.; Weber, A.M.; Jun, Y.; Zhu, Q.; Weisburd, B.; Huang, Y.; Audano, P.A.; Wang, H. Expectations and blind spots for structural variation detection from long-read assemblies and short-read genome sequencing technologies. Am. J. Hum. Genet. 2021, 108, 919–928. [Google Scholar] [CrossRef]
- Katsanis, S.H.; Katsanis, N. Molecular genetic testing and the future of clinical genomics. Nat. Rev. Genet. 2013, 14, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The third revolution in sequencing technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Sushmitha, T.J.; Rajeev, M.; Pandian, S.K. Next Generation Sequencing Advances and Applications in the World of Bacterial Diversity. In Microbial Systematics; CRC Press: Boca Raton, FL, USA, 2020; pp. 178–209. [Google Scholar]
- Iqbal, M.A.; Broeckel, U.; Levy, B.; Skinner, S.; Sahajpal, N.S.; Rodriguez, V.; Stence, A.; Awayda, K.; Scharer, G.; Skinner, C. Multisite assessment of optical genome mapping for analysis of structural variants in constitutional postnatal cases. J. Mol. Diagn. 2023, 25, 175–188. [Google Scholar] [CrossRef]
- Shree, A.; Shukla, P.C. Intellectual Disability: Definition, classification, causes and characteristics. Learn. Community Int. J. Educ. Soc. Dev. 2016, 7, 9–20. [Google Scholar] [CrossRef]
- Pyronneau, A.; He, Q.; Hwang, J.; Porch, M.; Contractor, A.; Zukin, R.S. Aberrant Rac1-cofilin signaling mediates defects in dendritic spines, synaptic function, and sensory perception in fragile X syndrome. Sci. Signal 2017, 10, eaan852. [Google Scholar] [CrossRef]
- Landucci, E.; Brindisi, M.; Bianciardi, L.; Catania, L.M.; Daga, S.; Croci, S.; Frullanti, E.; Fallerini, C.; Butini, S.; Brogi, S. iPSC-derived neurons profiling reveals GABAergic circuit disruption and acetylated α-tubulin defect which improves after iHDAC6 treatment in Rett syndrome. Exp. Cell Res. 2018, 368, 225–235. [Google Scholar] [CrossRef]
- D’Mello, S.R. Rett and Rett-related disorders: Common mechanisms for shared symptoms? Exp. Biol. Med. 2023, 248, 2095–2108. [Google Scholar] [CrossRef]
- Sweatt, J.D. Pitt–Hopkins syndrome: Intellectual disability due to loss of TCF4-regulated gene transcription. Exp. Mol. Med. 2013, 45, e21. [Google Scholar] [CrossRef]
- Gabriele, M.; Vulto-van Silfhout, A.T.; Germain, P.; Vitriolo, A.; Kumar, R.; Douglas, E.; Haan, E.; Kosaki, K.; Takenouchi, T.; Rauch, A. YY1 haploinsufficiency causes an intellectual disability syndrome featuring transcriptional and chromatin dysfunction. Am. J. Hum. Genet. 2017, 100, 907–925. [Google Scholar] [CrossRef]
- Rauen, K.A.; Tidyman, W.E. RASopathies—What they reveal about RAS/MAPK signaling in skeletal muscle development. Dis. Model. Mech. 2024, 17, dmm050609. [Google Scholar] [CrossRef]
- Borrie, S.C.; Brems, H.; Legius, E.; Bagni, C. Cognitive dysfunctions in intellectual disabilities: The contributions of the Ras-MAPK and PI3K-AKT-mTOR pathways. Annu. Rev. Genom. Hum. Genet. 2017, 18, 115–142. [Google Scholar] [CrossRef] [PubMed]
- Ba, W.; van der Raadt, J.; Kasri, N.N. Rho GTPase signaling at the synapse: Implications for intellectual disability. Exp. Cell Res. 2013, 319, 2368–2374. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, K.; Nalla, L.V.; Naresh, D.; Venkateswarlu, K.; Viswanadh, M.K.; Nalluri, B.N.; Chakravarthy, G.; Duguluri, S.; Singh, P.; Rai, S.N. WNT-β catenin signaling as a potential therapeutic target for neurodegenerative diseases: Current status and future perspective. Diseases 2023, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Kwan, V.; Unda, B.K.; Singh, K.K. Wnt signaling networks in autism spectrum disorder and intellectual disability. J. Neurodev. Disord. 2016, 8, 45. [Google Scholar] [CrossRef]
- Kessi, M.; Chen, B.; Peng, J.; Yan, F.; Yang, L.; Yin, F. Calcium channelopathies and intellectual disability: A systematic review. Orphanet J. Rare Dis. 2021, 16, 219. [Google Scholar] [CrossRef]
- Kessi, M.; Chen, B.; Peng, J.; Tang, Y.; Olatoutou, E.; He, F.; Yang, L.; Yin, F. Intellectual disability and potassium channelopathies: A systematic review. Front Genet 2020, 11, 614. [Google Scholar] [CrossRef]
- Liu, P.P.; Dai, S.K.; Mi, T.W.; Tang, G.B.; Wang, Z.; Wang, H.; Du, H.Z.; Tang, Y.; Teng, Z.Q.; Liu, C.M. Acetate supplementation restores cognitive deficits caused by ARID1A haploinsufficiency in excitatory neurons. Embo Mol. Med. 2022, 14, e15795. [Google Scholar] [CrossRef]
- Mir, Y.R.; Kuchay, R. Advances in identification of genes involved in autosomal recessive intellectual disability: A brief review. J. Med. Genet. 2019, 56, 567–573. [Google Scholar] [CrossRef]
- Torres, M.D.; Garcia, O.; Tang, C.; Busciglio, J. Dendritic spine pathology and thrombospondin-1 deficits in Down syndrome. Free Radic. Bio. Med. 2018, 114, 10–14. [Google Scholar] [CrossRef]
- Ruiz-Mejias, M.; de Lagran, M.M.; Mattia, M.; Castano-Prat, P.; Perez-Mendez, L.; Ciria-Suarez, L.; Gener, T.; Sancristobal, B.; García-Ojalvo, J.; Gruart, A. Overexpression of Dyrk1A, a Down syndrome candidate, decreases excitability and impairs gamma oscillations in the prefrontal cortex. J. Neurosci. 2016, 36, 3648–3659. [Google Scholar] [CrossRef]
- de San Martin, J.Z.; Delabar, J.; Bacci, A.; Potier, M. GABAergic over-inhibition, a promising hypothesis for cognitive deficits in Down syndrome. Free Radic. Bio. Med. 2018, 114, 33–39. [Google Scholar] [CrossRef]
- Pourhamzeh, M.; Moravej, F.G.; Arabi, M.; Shahriari, E.; Mehrabi, S.; Ward, R.; Ahadi, R.; Joghataei, M.T. The roles of serotonin in neuropsychiatric disorders. Cell Mol. Neurobiol. 2022, 42, 1671–1692. [Google Scholar] [CrossRef]
- Naddafi, F.; Mirshafiey, A. The neglected role of histamine in Alzheimer’s disease. Am. J. Alzheimer’s Dis. Other Dement. 2013, 28, 327–336. [Google Scholar] [CrossRef]
- Miclea, D.; Peca, L.; Cuzmici, Z.; Pop, I.V. Genetic testing in patients with global developmental delay/intellectual disabilities. A review. Clujul Med. 2015, 88, 288. [Google Scholar] [CrossRef]
- Ozawa, T.; Ishihara, S.; Kawai, K.; Kazama, S.; Yamaguchi, H.; Sunami, E.; Kitayama, J.; Watanabe, T. Impact of a lymphocyte to monocyte ratio in stage IV colorectal cancer. J. Surg. Res. 2015, 199, 386–392. [Google Scholar] [CrossRef]
- Vatsa, N.; Jana, N.R. UBE3A and its link with autism. Front. Mol. Neurosci. 2018, 11, 448. [Google Scholar] [CrossRef]
- Margolis, S.S.; Sell, G.L.; Zbinden, M.A.; Bird, L.M. Angelman syndrome. Neurotherapeutics 2015, 12, 641–650. [Google Scholar] [CrossRef]
- Khatri, N. The Autism Protein UBE3A/E6AP Regulates Remodeling of Neuronal Dendritic Arborization; Boston University: Boston, MA, USA, 2017. [Google Scholar]
- Kim, H.; Kunz, P.A.; Mooney, R.; Philpot, B.D.; Smith, S.L. Maternal loss of Ube3a impairs experience-driven dendritic spine maintenance in the developing visual cortex. J. Neurosci. 2016, 36, 4888–4894. [Google Scholar] [CrossRef]
- Wieczorek, D. Autosomal dominant intellectual disability. Med. Genet. 2018, 30, 318–322. [Google Scholar] [CrossRef]
- Marco, S.S.; Pisón, J.L.; Escribano, C.C.; Viejo, I.G.; Gallart, M.M.; Villagrasa, P.S. Neurological manifestations of neurofibromatosis type 1: Our experience. Neurología 2022, 37, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Khan, S.; Windpassinger, C.; Badar, M.; Nawaz, Z.; Mohammad, R.M. The molecular genetics of autosomal recessive nonsyndromic intellectual disability: A mutational continuum and future recommendations. Ann. Hum. Genet. 2016, 80, 342–368. [Google Scholar] [CrossRef]
- Iroegbu, J.D.; Ijomone, O.K.; Femi-Akinlosotu, O.M.; Ijomone, O.M. ERK/MAPK signalling in the developing brain: Perturbations and consequences. Neurosci. Biobehav. Rev. 2021, 131, 792–805. [Google Scholar] [CrossRef] [PubMed]
- Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallée, B.; Bénédetti, H. Neurofibromin structure, functions and regulation. Cells 2020, 9, 2365. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, M.; Johnston, M.V.; Stafstrom, C.E. SYNGAP1 mutations: Clinical, genetic, and pathophysiological features. Int. J. Dev. Neurosci. 2019, 78, 65–76. [Google Scholar] [CrossRef]
- Araki, Y.; Hong, I.; Gamache, T.R.; Ju, S.; Collado-Torres, L.; Shin, J.H.; Huganir, R.L. SynGAP isoforms differentially regulate synaptic plasticity and dendritic development. eLife 2020, 9, e56273. [Google Scholar] [CrossRef]
- Kilinc, M.; Arora, V.; Creson, T.K.; Rojas, C.; Le, A.A.; Lauterborn, J.; Wilkinson, B.; Hartel, N.; Graham, N.; Reich, A. Endogenous Syngap1 alpha splice forms promote cognitive function and seizure protection. eLife 2022, 11, e75707. [Google Scholar] [CrossRef]
- Canela, N.; Rodriguez-Vilarrupla, A.; Estanyol, J.M.; Díaz, C.; Pujol, M.J.; Agell, N.; Bachs, O. The SET protein regulates G2/M transition by modulating cyclin B-cyclin-dependent kinase 1 activity. J. Biol. Chem. 2003, 278, 1158–1164. [Google Scholar] [CrossRef]
- Stevens, S.J.; van der Schoot, V.; Leduc, M.S.; Rinne, T.; Lalani, S.R.; Weiss, M.M.; van Hagen, J.M.; Lachmeijer, A.M.; Study, C.; Stockler Ipsiroglu, S.G. De novo mutations in the SET nuclear proto-oncogene, encoding a component of the inhibitor of histone acetyltransferases (INHAT) complex in patients with nonsyndromic intellectual disability. Hum. Mutat. 2018, 39, 1014–1023. [Google Scholar] [CrossRef]
- Pan, X.; Liu, S.; Feng, X.; Liu, L.; Zhang, X.; Qian, G.; Liang, N.; Yao, H.; Dong, X.; Tan, B. Whole exome sequencing and transcriptome analysis in two unrelated patients with novel SET mutations. J. Hum. Genet. 2023, 68, 867–874. [Google Scholar] [CrossRef]
- Kon, N.; Wang, D.; Gu, W. Loss of SET reveals both the p53-dependent and the p53-independent functions in vivo. Cell Death Dis. 2019, 10, 237. [Google Scholar] [CrossRef] [PubMed]
- Bauman, M.L.; Kemper, T.L. Morphologic and histoanatomic observations of the brain in untreated human phenylketonuria. Acta Neuropathol. 1982, 58, 55–63. [Google Scholar] [CrossRef]
- Winn, S.R.; Scherer, T.; Thöny, B.; Harding, C.O. High dose sapropterin dihydrochloride therapy improves monoamine neurotransmitter turnover in murine phenylketonuria (PKU). Mol. Genet. Metab. 2016, 117, 5–11. [Google Scholar] [CrossRef]
- McKean, C.M. The effects of high phenylalanine concentrations on serotinin and catecholamine metabolism in the human brain. Brain Res. 1972, 47, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, D.; Bruinenberg, V.M.; Mazzola, P.N.; Van Faassen, M.H.; De Blaauw, P.; Pascucci, T.; Puglisi-Allegra, S.; Kema, I.P.; Heiner-Fokkema, M.R.; Van Der Zee, E.A. Therapeutic brain modulation with targeted large neutral amino acid supplements in the Pah-enu2 phenylketonuria mouse model. Am. J. Clin. Nutr. 2016, 104, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Bilder, D.A.; Noel, J.K.; Baker, E.R.; Irish, W.; Chen, Y.; Merilainen, M.J.; Prasad, S.; Winslow, B.J. Systematic review and meta-analysis of neuropsychiatric symptoms and executive functioning in adults with phenylketonuria. Dev. Neuropsychol. 2016, 41, 245–260. [Google Scholar] [CrossRef]
- Jahja, R.; van Spronsen, F.J.; De Sonneville, L.M.; van der Meere, J.J.; Bosch, A.M.; Hollak, C.E.; Rubio-Gozalbo, M.E.; Brouwers, M.C.; Hofstede, F.C.; de Vries, M.C. Long-term follow-up of cognition and mental health in adult phenylketonuria: A PKU-COBESO study. Behav. Genet. 2017, 47, 486–497. [Google Scholar] [CrossRef]
- Usman, M.; Li, Y.; Ke, Y.; Chhetri, G.; Islam, M.A.; Wang, Z.; Li, X. Trappc9 deficiency impairs the plasticity of stem cells. Int. J. Mol. Sci. 2022, 23, 4900. [Google Scholar] [CrossRef]
- Marangi, G.; Leuzzi, V.; Manti, F.; Lattante, S.; Orteschi, D.; Pecile, V.; Neri, G.; Zollino, M. TRAPPC9-related autosomal recessive intellectual disability: Report of a new mutation and clinical phenotype. Eur. J. Hum. Genet. 2013, 21, 229–232. [Google Scholar] [CrossRef]
- Park, C.; Lee, S.; Yoon, K. Epitranscriptomic regulation of transcriptome plasticity in development and diseases of the brain. Bmb Rep. 2020, 53, 551. [Google Scholar] [CrossRef]
- Komara, M.; Al-Shamsi, A.M.; Ben-Salem, S.; Ali, B.R.; Al-Gazali, L. A novel single-nucleotide deletion (c. 1020delA) in NSUN2 causes intellectual disability in an emirati child. J. Mol. Neurosci. 2015, 57, 393–399. [Google Scholar] [CrossRef]
- Martin, J.P.; Bell, J. A pedigree of mental defect showing sex-linkage. J. Neurol. Psychiatry 1943, 6, 154. [Google Scholar] [CrossRef] [PubMed]
- Flore, L.A.; Milunsky, J.M. Updates in the genetic evaluation of the child with global developmental delay or intellectual disability. Semin. Pediatr. Neurol. 2012, 19, 173–180. [Google Scholar] [CrossRef]
- Saldarriaga, W.; Tassone, F.; González-Teshima, L.Y.; Forero-Forero, J.V.; Ayala-Zapata, S.; Hagerman, R. Fragile X syndrome. Colomb. Medica 2014, 45, 190–198. [Google Scholar] [CrossRef]
- American Psychiatric Association, D.S. Diagnostic and Statistical Manual of Mental Disorders: DSM-5; American Psychiatric Association: Washington, DC, USA, 2013; Volume 5. [Google Scholar]
- Parsamanesh, N.; Miri-Moghaddam, E. Novel insight into intellectual disability: A review article. Gene Cell Tissue 2018, 5, e84401. [Google Scholar] [CrossRef]
- Gabis, L.V.; Shaham, M.; Attia, O.L.; Kowal, T.; David, S.; Banet-Levi, Y.; Shefer, S.; Gabis, D.; Mula-Topf, D.; Avrech Bar, M. An escalating continuum of learning and attention difficulties from premutation to full mutation in female carriers of FMR1 expansion. Front. Neurol. 2023, 14, 1135630. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 1–19. [Google Scholar] [CrossRef]
- Shitik, E.M.; Velmiskina, A.A.; Dolskiy, A.A.; Yudkin, D.V. Reactivation of FMR1 gene expression is a promising strategy for fragile X syndrome therapy. Gene Ther. 2020, 27, 247–253. [Google Scholar] [CrossRef]
- Bassell, G.J.; Warren, S.T. Fragile X syndrome: Loss of local mRNA regulation alters synaptic development and function. Neuron 2008, 60, 201–214. [Google Scholar] [CrossRef]
- Chen, Y.S.; Guo, L.; Han, M.; Zhang, S.M.; Chen, Y.Q.; Zou, J.; Bai, S.Y.; Cheng, G.R.; Zeng, Y. Cerebellum neuropathology and motor skill deficits in fragile X syndrome. Int. J. Dev. Neurosci. 2022, 82, 557–568. [Google Scholar] [CrossRef]
- Lyst, M.J.; Bird, A. Rett syndrome: A complex disorder with simple roots. Nat. Rev. Genet. 2015, 16, 261–275. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, S.R., III. MECP2 and the biology of MECP2 duplication syndrome. J. Neurochem. 2021, 159, 29–60. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, V.R.; Rastegar, M. Rett syndrome and MeCP2. Neuromol. Med. 2014, 16, 231–264. [Google Scholar] [CrossRef]
- Ip, J.P.; Mellios, N.; Sur, M. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat. Rev. Neurosci. 2018, 19, 368–382. [Google Scholar] [CrossRef]
- van Schalkwyk, G.I.; Lewis, A.S.; Beyer, C.; Johnson, J.; van Rensburg, S.; Bloch, M.H. Efficacy of antipsychotics for irritability and aggression in children: A meta-analysis. Expert. Rev. Neurother. 2017, 17, 1045–1053. [Google Scholar] [CrossRef]
- Alsayouf, H.A.; Talo, H.; Biddappa, M.L.; De Los Reyes, E. Risperidone or aripiprazole can resolve autism core signs and symptoms in young children: Case study. Children 2021, 8, 318. [Google Scholar] [CrossRef] [PubMed]
- Politte, L.C.; McDougle, C.J. Atypical antipsychotics in the treatment of children and adolescents with pervasive developmental disorders. Psychopharmacology 2014, 231, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Matson, J.L.; Mahan, S. Antipsychotic drug side effects for persons with intellectual disability. Res. Dev. Disabil. 2010, 31, 1570–1576. [Google Scholar] [CrossRef]
- McCarty, D.M.; Fu, H. Gene Therapy for Neurodevelopmental Disorders. In Encyclopedia of Life Sciences; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Li, G.; Li, X.; Zhuang, S.; Wang, L.; Zhu, Y.; Chen, Y.; Sun, W.; Wu, Z.; Zhou, Z.; Chen, J. Gene editing and its applications in biomedicine. Sci. China Life Sci. 2022, 65, 660–700. [Google Scholar] [CrossRef]
- Ilyas, M.; Mir, A.; Efthymiou, S.; Houlden, H. The genetics of intellectual disability: Advancing technology and gene editing. F1000Research 2020, 9, 22. [Google Scholar] [CrossRef]
- Turner, T.J.; Zourray, C.; Schorge, S.; Lignani, G. Recent advances in gene therapy for neurodevelopmental disorders with epilepsy. J. Neurochem. 2021, 157, 229–262. [Google Scholar] [CrossRef]
- McCamphill, P.K.; Stoppel, L.J.; Senter, R.K.; Lewis, M.C.; Heynen, A.J.; Stoppel, D.C.; Sridhar, V.; Collins, K.A.; Shi, X.; Pan, J.Q. Selective inhibition of glycogen synthase kinase 3α corrects pathophysiology in a mouse model of fragile X syndrome. Sci. Transl. Med. 2020, 12, eaam8572. [Google Scholar] [CrossRef] [PubMed]
- Rath, J. Safety and security risks of CRISPR/Cas9. In Ethics Dumping: Case Studies from North-South Research Collaborations; Springer: Cham, Switzerland, 2018; pp. 107–113. [Google Scholar]
- Bennett, C.F. Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Milazzo, C.; Mientjes, E.J.; Wallaard, I.; Rasmussen, S.V.; Erichsen, K.D.; Kakunuri, T.; van der Sman, A.E.; Kremer, T.; Miller, M.T.; Hoener, M.C. Antisense oligonucleotide treatment rescues UBE3A expression and multiple phenotypes of an Angelman syndrome mouse model. JCI Insight 2021, 6, e145991. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Ward, A.J.; Chun, S.; Bennett, C.F.; Beaudet, A.L.; Rigo, F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 2015, 518, 409–412. [Google Scholar] [CrossRef]
- Pysh, J.J.; Weiss, G.M. Exercise during development induces an increase in Purkinje cell dendritic tree size. Science 1979, 206, 230–232. [Google Scholar] [CrossRef]
- Sahnoune, I.; Inoue, T.; Kesler, S.R.; Rodgers, S.P.; Sabek, O.M.; Pedersen, S.E.; Zawaski, J.A.; Nelson, K.H.; Ris, M.D.; Leasure, J.L. Exercise ameliorates neurocognitive impairments in a translational model of pediatric radiotherapy. Neuro Oncol. 2018, 20, 695–704. [Google Scholar] [CrossRef]
- Stranahan, A.M.; Khalil, D.; Gould, E. Running induces widespread structural alterations in the hippocampus and entorhinal cortex. Hippocampus 2007, 17, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Bonsale, R.; Infantino, R.; Perrone, M.; Marabese, I.; Ricciardi, F.; Fusco, A.; Teweldemedhin, M.M.; Boccella, S.; Guida, F.; Rinaldi, B. The long-term exercise after traumatic brain injury: Reharmonizing brain by sound body. Brain Res. 2023, 1816, 148471. [Google Scholar] [CrossRef]
- Mu, L.; Cai, J.; Gu, B.; Yu, L.; Li, C.; Liu, Q.; Zhao, L. Treadmill exercise prevents decline in spatial learning and memory in 3× Tg-AD mice through enhancement of structural synaptic plasticity of the hippocampus and prefrontal cortex. Cells 2022, 11, 244. [Google Scholar] [CrossRef]
- Schneider, S.; Vogt, T.; Frysch, J.; Guardiera, P.; Strüder, H.K. School sport—A neurophysiological approach. Neurosci. Lett. 2009, 467, 131–134. [Google Scholar] [CrossRef]
- Chaddock-Heyman, L.; Erickson, K.I.; Chappell, M.A.; Johnson, C.L.; Kienzler, C.; Knecht, A.; Drollette, E.S.; Raine, L.B.; Scudder, M.R.; Kao, S. Aerobic fitness is associated with greater hippocampal cerebral blood flow in children. Dev. Cogn. Neuros-Neth. 2016, 20, 52–58. [Google Scholar] [CrossRef]
- Krafft, C.E.; Pierce, J.E.; Schwarz, N.F.; Chi, L.; Weinberger, A.L.; Schaeffer, D.J.; Rodrigue, A.L.; Camchong, J.; Allison, J.D.; Yanasak, N.E. An eight month randomized controlled exercise intervention alters resting state synchrony in overweight children. Neuroscience 2014, 256, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Cornejo, I.; Cadenas-Sanchez, C.; Contreras-Rodriguez, O.; Verdejo-Roman, J.; Mora-Gonzalez, J.; Migueles, J.H.; Henriksson, P.; Davis, C.L.; Verdejo-Garcia, A.; Catena, A. A whole brain volumetric approach in overweight/obese children: Examining the association with different physical fitness components and academic performance. The ActiveBrains project. Neuroimage 2017, 159, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Belcher, B.R.; Zink, J.; Azad, A.; Campbell, C.E.; Chakravartti, S.P.; Herting, M.M. The roles of physical activity, exercise, and fitness in promoting resilience during adolescence: Effects on mental well-being and brain development. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2021, 6, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Vancampfort, D.; Van Damme, T.; Firth, J.; Stubbs, B.; Schuch, F.; Suetani, S.; Arkesteyn, A.; Van Biesen, D. Physical activity correlates in children and adolescents, adults, and older adults with an intellectual disability: A systematic review. Disabil. Rehabil. 2022, 44, 4189–4200. [Google Scholar] [CrossRef] [PubMed]
- Bouzas, S.; Martínez-Lemos, R.I.; Ayan, C. Effects of exercise on the physical fitness level of adults with intellectual disability: A systematic review. Disabil. Rehabil. 2019, 41, 3118–3140. [Google Scholar] [CrossRef]
- Axmon, A.; Ahlström, G.; Sandberg, M. Falls resulting in health care among older people with intellectual disability in comparison with the general population. J. Intell. Disabil. Res. 2019, 63, 193–204. [Google Scholar] [CrossRef]
- Li, X.; Huang, J.; Kong, Z.; Sun, F.; Sit, C.H.; Li, C. Effects of virtual reality-based exercise on physical fitness in people with intellectual disability: A systematic review of randomized controlled trials. Games Health J. 2023, 12, 89–99. [Google Scholar] [CrossRef]
- Pastula, R.M.; Stopka, C.B.; Delisle, A.T.; Hass, C.J. Effect of moderate-intensity exercise training on the cognitive function of young adults with intellectual disabilities. J. Strength Cond. Res. 2012, 26, 3441–3448. [Google Scholar] [CrossRef]
- Vogt, T.; Schneider, S.; Anneken, V.; Strüder, H.K. Moderate cycling exercise enhances neurocognitive processing in adolescents with intellectual and developmental disabilities. Res. Dev. Disabil. 2013, 34, 2708–2716. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Brümmer, V.; Abel, T.; Askew, C.D.; Strüder, H.K. Changes in brain cortical activity measured by EEG are related to individual exercise preferences. Physiol. Behav. 2009, 98, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Vogt, T.; Schneider, S.; Abeln, V.; Anneken, V.; Strüder, H.K. Exercise, mood and cognitive performance in intellectual disability—A neurophysiological approach. Behav. Brain Res. 2012, 226, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Carmeli, E.; Zinger-Vaknin, T.; Morad, M.; Merrick, J. Can physical training have an effect on well-being in adults with mild intellectual disability? Mech. Ageing Dev. 2005, 126, 299–304. [Google Scholar] [CrossRef]
- Cantelon, J.A.; Giles, G.E. A review of cognitive changes during acute aerobic exercise. Front. Psychol. 2021, 12, 653158. [Google Scholar] [CrossRef]
- Vissers, L.E.; Gilissen, C.; Veltman, J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2016, 17, 9–18. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Name | Catalog | Advantages | Disadvantages |
|---|---|---|---|
| Sanger sequencing | A long sequencing length (1 kb) and a high accuracy rate of 100% | Time consuming (weeks or even months of work) and expensive (millions of dollars), requires known genomic loc | |
| Next-generation Sequencing (NGS) | Whole-exome sequencing (WES) | Short turnaround time (only days or weeks of work), low cost (thousands of dollars), and high diagnostic rate (millions of reads) | Can only search DNA sequences in the exonic regions |
| Whole-genome sequencing (WGS) | Captures both exonic and intronic regions, identifies pathogenic variations in complex genes, and improves de novo mutations detection | Short reads (400 bp or 2 × 300 bp) | |
| Third-generation sequencing (TGS) | Overcomes the biases-related issues introduced by PCR amplification and dephasing and increases the length of base reads | High error rate during sequencing (an error rate of 10–20%) |
| Gene | Pathogenesis | Phenotype |
|---|---|---|
| MECP2 | abnormal transcriptional regulation and chromatin remodeling | Immature and decreased synapse numbers Impaired synaptic plasticity (e.g., LTP ↓) Decreased dendritic density Dendritic spine immature and decreased spine numbers |
| UBE3A | affecting the ubiquitin-proteasome pathway | |
| DYRK1A | abnormal encode a kinase involved in brain development | |
| PTEN | dysregulated cell growth and proliferation | |
| TCE4 | abnormal transcriptional regulation | |
| GNAS | abnormal G protein signaling | |
| CHD8 | disrupt chromatin remodeling and gene expression | |
| KDM5C | affect a neurodevelopmental kinase | |
| PHF | neurodevelopmental gene expression dysregulation | |
| EHMT1 | neurodevelopmental gene expression dysregulation | |
| NF1 | dysregulated cell growth and proliferation | |
| ZDHHC9 | abnormal protein palmitoylation | |
| ATRX | chromosomal abnormalities and defective DNA repair | |
| FOXP1 | abnormal transcriptional regulation | |
| FMR1 | impaired FMRP protein production | |
| TSC1/TSC2 | abnormal cell growth and proliferation | |
| CDKL5 | affect a neurodevelopmental kinase | |
| TRAPPC9 | affects nerve growth factor-induced neuronal differentiation | |
| USUN2 | decreased tRNA methyltransferase function | |
| SHANK3 | abnormal postsynaptic protein density | Impaired synaptic plasticity (e.g., LTP↓) Abnormal neuronal signaling |
| SYNGAP1 | de novo variations in certain proteins | |
| NRXN1 | abnormal presynaptic protein synthesis | |
| SCN2A | sodium channel dysfunction | |
| CACNA1A | calcium channel dysfunction | |
| STXBP1 | dysfunction of neurotransmitter release process | |
| GRIN2B | NMDA receptor dysfunction | |
| GABRA1 | GABA receptor dysfunction |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, K.; Zheng, X. A 10-Year Review on Advancements in Identifying and Treating Intellectual Disability Caused by Genetic Variations. Genes 2024, 15, 1118. https://doi.org/10.3390/genes15091118
Hou K, Zheng X. A 10-Year Review on Advancements in Identifying and Treating Intellectual Disability Caused by Genetic Variations. Genes. 2024; 15(9):1118. https://doi.org/10.3390/genes15091118
Chicago/Turabian StyleHou, Kexin, and Xinyan Zheng. 2024. "A 10-Year Review on Advancements in Identifying and Treating Intellectual Disability Caused by Genetic Variations" Genes 15, no. 9: 1118. https://doi.org/10.3390/genes15091118