
Genetic Foundations of Nellore Traits: A Gene Prioritization and Functional Analyses of Genome-Wide Association Study Results


Abstract
1. Introduction
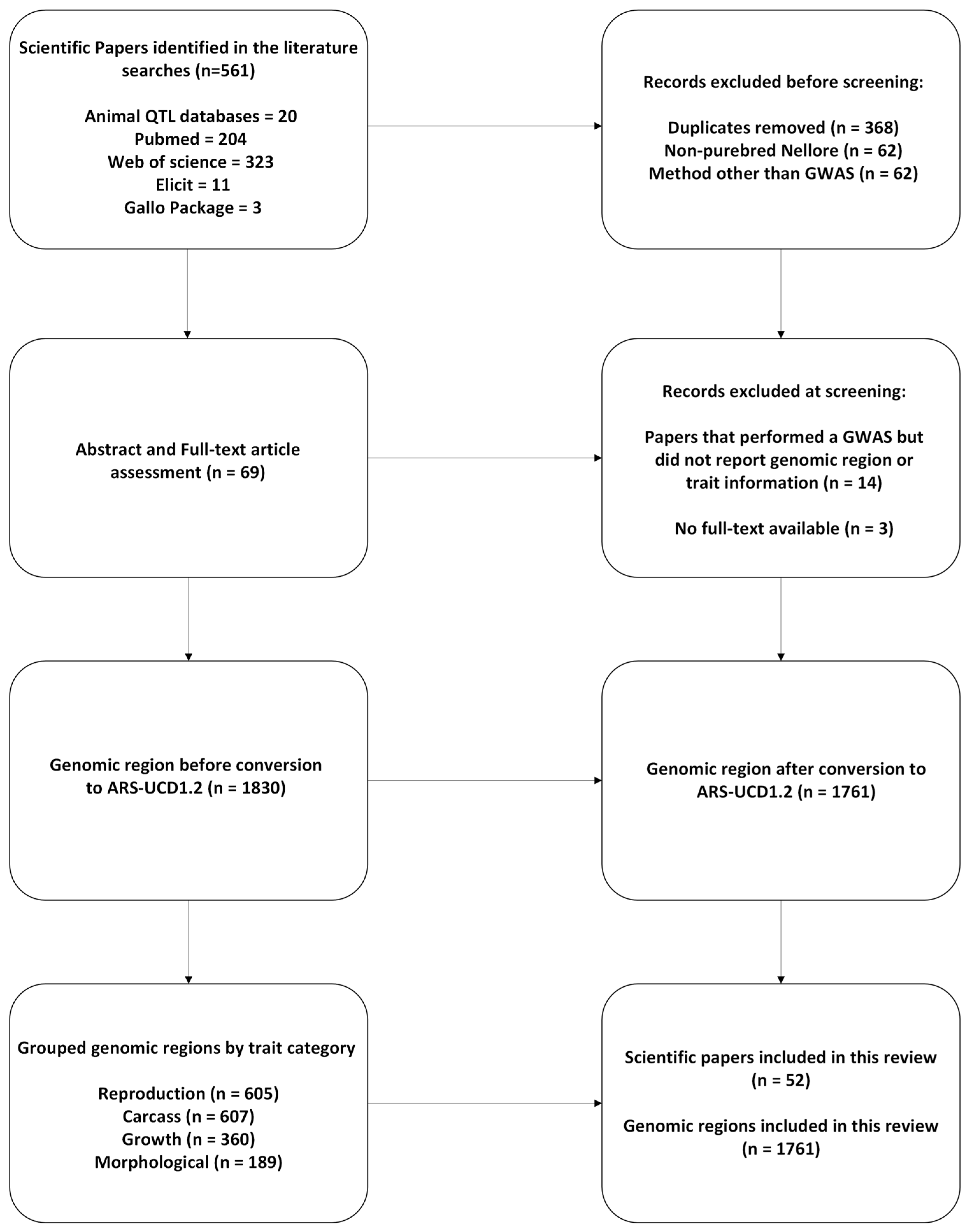
2. Materials and Methods
2.1. Data Gathering and Editing
2.2. Trait Groups
- Reproduction traits: scrotal circumference, age at puberty, early pregnancy, early puberty, precocity, age at first calving, heifer rebreeding, number of calving, testicular hypoplasia, gestation length, preweaning calf mortality rate, antral follicle count, stayability, anti-Müllerian hormone, gestation length, calving interval, calving ease, and post-natal mortality.
- 1.a.
- Subgroup: Sexual precocityIncluded traits: scrotal circumference, early pregnancy, early puberty, age at first calving, antral follicle count, calving interval, and calving ease.
- Carcass traits: backfat thickness, rib eye area, rump fat thickness, hot carcass weight, subcutaneous fat thickness, intramuscular fat content, longissimus muscle area, marbling, tenderness, shear force tenderness, and meat color.
- 2.a.
- Subgroup: Meat qualityIncluded traits: marbling, tenderness, shear force tenderness, and meat color.
- Growth-related traits: residual body weight gain, average daily gain, accumulated productivity, birth weight, weaning weight, yearling weight, adult cow weight, weight gain from birth to weaning, weight gain from weaning to yearling, yearling height, residual feed intake, dry matter intake, feed efficiency, and feed conversion ratio.
- 3.a.
- Subgroup: EfficiencyIncluded traits: residual feed intake, dry matter intake, feed efficiency, and feed conversion ratio.
- Morphological traits: body conformation, muscularity, precocity, feet and leg deformation, and feet and leg conformation.
- 4.a.
- Subgroup: Visual scoresIncluded traits: conformation, precocity, and muscling.
2.3. Over-Representation and Prioritization Analyses
- Reproduction and sexual precocity traits: “Fertility”, “implantation”, “preimplantation”, “endometrium”, “embryonic development”, “primordial follicles”, “uterus”, “luteal”, “gestation”, “embryo”, “ovulation”, “estrogen”, “estradiol”, “endocrine hormone”, “gamete, mammary glands”, “lactation”, “pregnancy”, “oocyte”, “fetus”, “zygote”, “ovary”, “amniotic”, “cervical mucosa follicle”, “Gonad”, “antral follicles”, “pre-eclampsia”, “placenta”, “testes”, “sperm”, “scrotal circumference”, “scrotal, testicular”, “testis”, “semen”, “spermatozoa”, “spermatogenesis”, “testicular hypoplasia”, and “hypogonadism”.
- Carcass and meat quality traits: “Longissimus muscle”, “Backfat thickness”, “Myosin”, “Actin”, “Fibroblast”, “Tissue deposition”, “Fat deposition”, “Muscle, Subcutaneous fat”, “Biceps femoris”, “Carcass weight”, and “Skeletal muscle”.
- Morphological and visual score traits: “body development”, “muscle mass”, “skeletal muscle”, “subcutaneous fat”, “conformational structure”, “adipose tissue”, “body depth”, “feet and leg”, “foot angle”, “tendon”, “joints”, “mobility aplomb”, “osteogenic differentiation”, “articular cartilage”, and “fat deposition”.
- Growth-related and efficiency traits: “body weight”, “birth weight”, “adult weight”, “weight gain”, “body size”, “average daily gain”, “body weight gain”, “daily gain”, “stature”, “growth”, “adipose tissue”, “dry matter intake”, “metabolism”, “maternal behavior”, “maternal ability”, “feed conversion”, “feed efficiency”, “dry matter”, “milk composition”, and “muscle development”.
2.4. Functional Analysis
2.5. Venn Diagrams and Gene Network Integration
3. Results and Discussion
3.1. Gene Annotation and Prioritization Analysis
3.2. Enrichment and Gene Integration Analysis of the Functional Candidate Genes Identified within Groups
3.2.1. Reproduction and Sexual Precocity Traits
3.2.2. Carcass and Meat Quality Traits
3.2.3. Growth-Related and Efficiency Traits
3.2.4. Morphological and Visual Score Traits
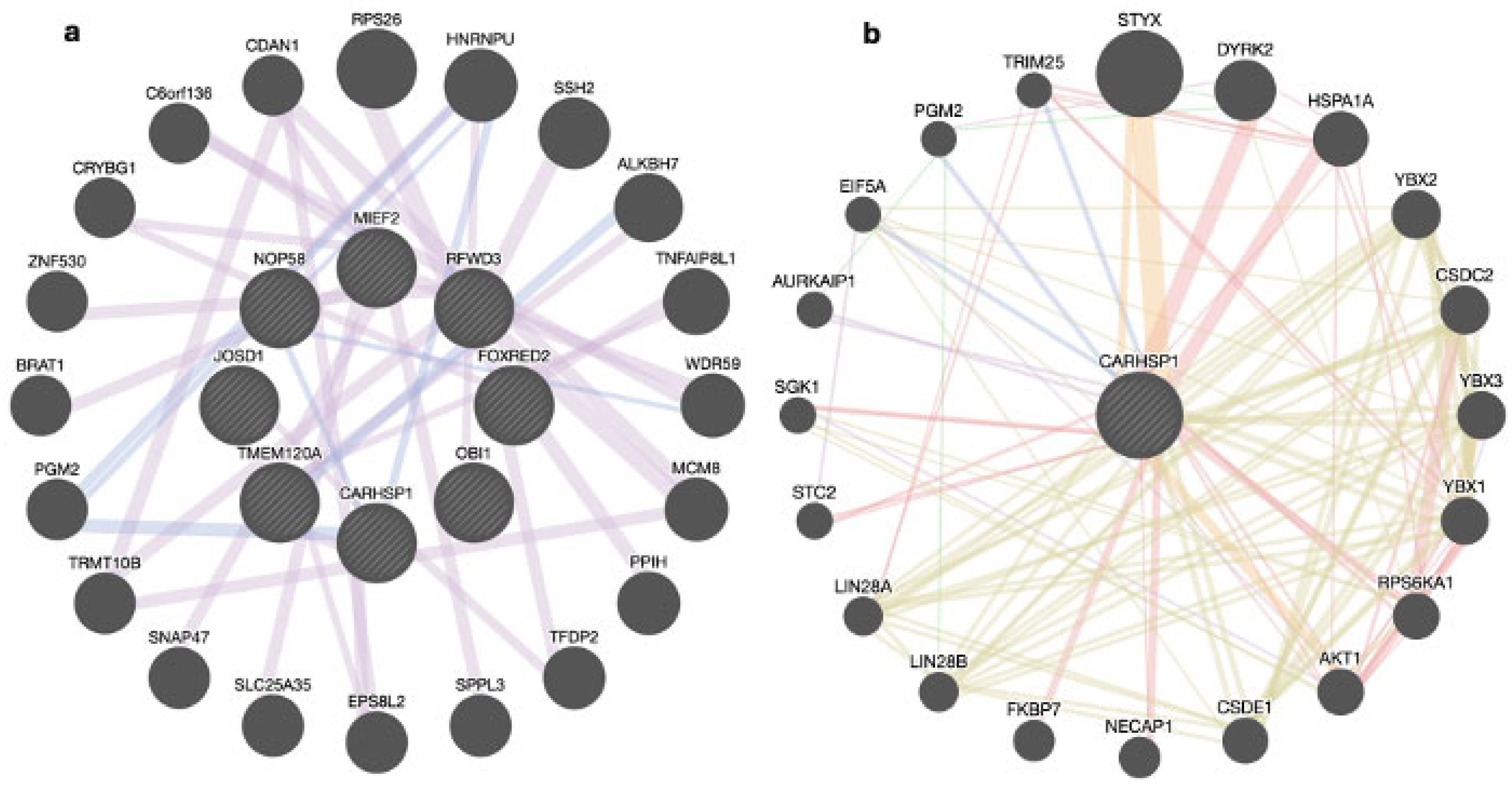
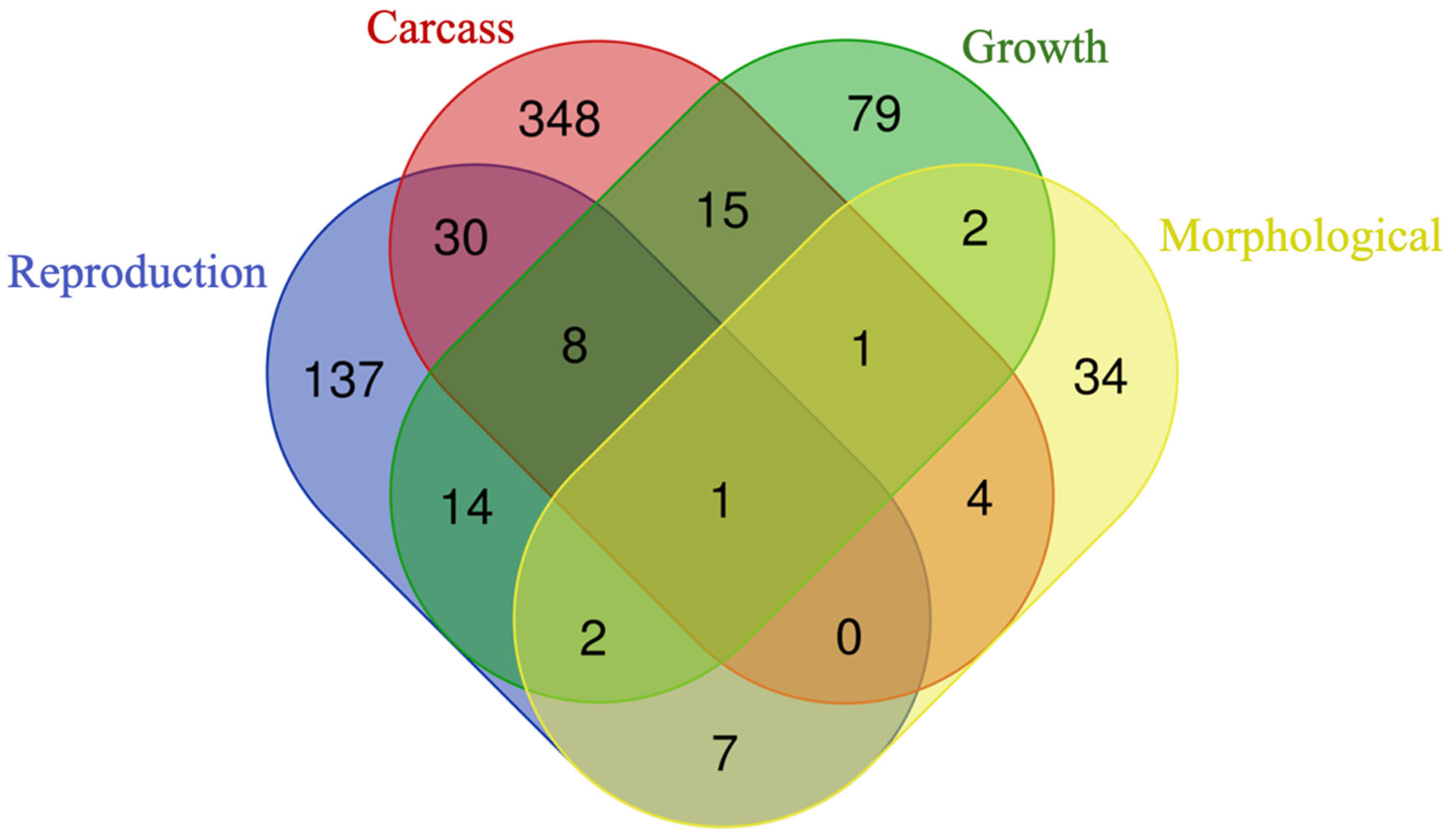
3.3. Genes Shared among Groups
3.3.1. Over-Represented Genes
3.3.2. Prioritized Genes
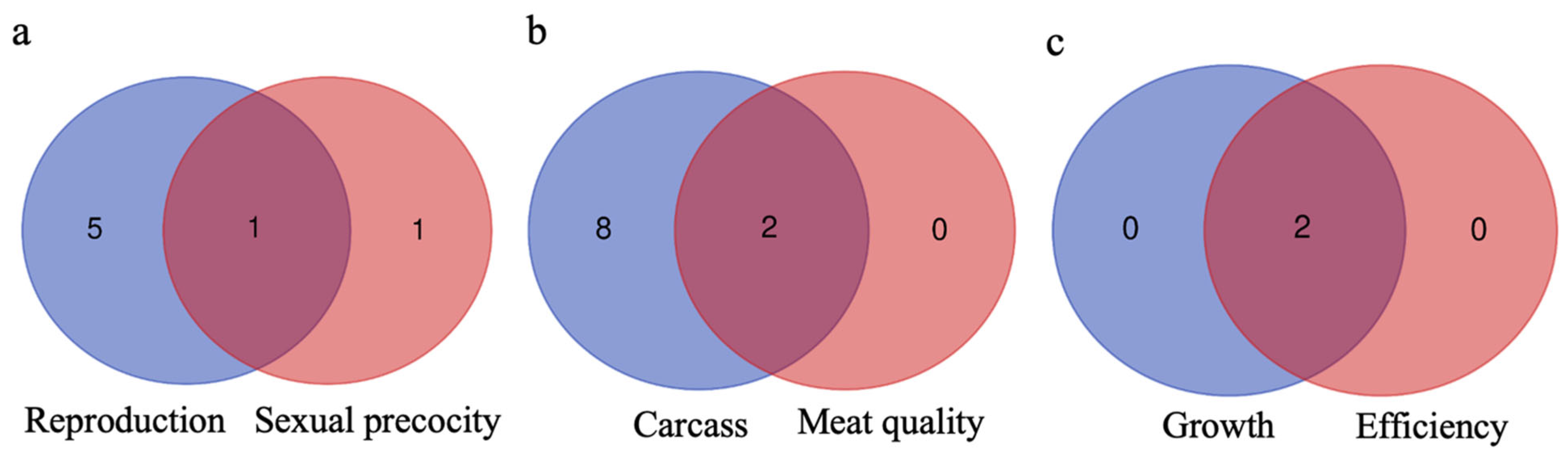
3.4. Genes Shared between Groups and Subgroups
4. Limitations of This Study and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Utsunomiya, Y.T.; Do Carmo, A.S.; Carvalheiro, R.; Neves, H.H.; Matos, M.C.; Zavarez, L.B.; Pérez O’Brien, A.M.; Sölkner, J.; McEwan, J.C.; Cole, J.B.; et al. Genome-wide association study for birth weight in Nellore cattle points to previously described orthologous genes affecting human and bovine height. BMC Genet. 2013, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Pitt, D.; Sevane, N.; Nicolazzi, E.L.; MacHugh, D.E.; Park, S.D.E.; Colli, L.; Martinez, R.; Bruford, M.W.; Orozco-terWengel, P. Domestication of cattle: Two or three events? Evol. Appl. 2018, 12, 123–136. [Google Scholar] [CrossRef]
- De Albuquerque, L.G. Proceedings of the 8th World Congress on Genetics Applied to Livestock Production, Belo Horizonte, Brazil, 13–18 August 2006. Instituto Prociência: San Borja, Peru, 2006. [Google Scholar]
- Porto-Neto, L.R.; Reverter, A.; Prayaga, K.C.; Chan, E.K.; Johnston, D.J.; Hawken, R.J.; Fordyce, G.; Garcia, J.F.; Sonstegard, T.S.; Bolormaa, S.; et al. The genetic architecture of climatic adaptation of tropical cattle. PLoS ONE 2014, 9, e113284. [Google Scholar] [CrossRef] [PubMed]
- Carvalheiro, R.; Boison, S.A.; Neves, H.H.; Sargolzaei, M.; Schenkel, F.S.; Utsunomiya, Y.T.; O’Brien, A.M.; Sölkner, J.; McEwan, J.C.; Van Tassell, C.P.; et al. Accuracy of genotype imputation in Nellore cattle. Genet. Sel. Evol. GSE 2014, 46, 69. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, L.; Fernandes Júnior, G.; Carvalheiro, R. Beef Cattle Genomic Selection In Tropical Environments. Proc. Assoc. Advmt. Anim. Breed. Genet. 2017, 22, 255–263. [Google Scholar]
- Fernandes Júnior, G.A.; Peripolli, E.; Schmidt, P.I.; Campos, G.S.; Mota, L.F.M.; Mercadante, M.E.Z.; Baldi, F.; Carvalheiro, R.; De Albuquerque, L.G. Current applications and perspectives of genomic selection in Bos indicus (Nellore) cattle. Livest. Sci. 2022, 263, 105001. [Google Scholar] [CrossRef]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef]
- Mokry, F.B.; Buzanskas, M.E.; De Alvarenga Mudadu, M.; Do Amaral Grossi, D.; Higa, R.H.; Ventura, R.V.; De Lima, A.O.; Sargolzaei, M.; Conceição Meirelles, S.L.; Schenkel, F.S.; et al. Linkage disequilibrium and haplotype block structure in a composite beef cattle breed. BMC Genom. 2014, 15, S6. [Google Scholar] [CrossRef]
- Sanchez, M.-P.; Tribout, T.; Kadri, N.K.; Chitneedi, P.K.; Maak, S.; Hozé, C.; Boussaha, M.; Croiseau, P.; Philippe, R.; Spengeler, M.; et al. Sequence-based GWAS meta-analyses for beef production traits. Genet. Sel. Evol. 2023, 55, 70. [Google Scholar] [CrossRef]
- Irano, N.; De Camargo, G.M.F.; Costa, R.B.; Terakado, A.P.N.; Magalhães, A.F.B.; Silva, R.M.D.O.; Dias, M.M.; Bignardi, A.B.; Baldi, F.; Carvalheiro, R.; et al. Genome-Wide Association Study for Indicator Traits of Sexual Precocity in Nellore Cattle. PLoS ONE 2016, 11, e0159502. [Google Scholar] [CrossRef]
- Silva, D.O.; Fernandes Júnior, G.A.; Fonseca, L.F.S.; Mota, L.F.M.; Bresolin, T.; Carvalheiro, R.; De Albuquerque, L.G. Genome-wide association study for stayability at different calvings in Nellore beef cattle. BMC Genom. 2024, 25, 93. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, F.E.; Espigolan, R.; Berton, M.P.; Neto, J.B.S.; Silva, R.P.; Grigoletto, L.; Silva, R.M.O.; Ferraz, J.B.S.; Eler, J.P.; Aguilar, I.; et al. Genome-wide association study and predictive ability for growth traits in Nellore cattle. Livest. Sci. 2020, 231, 103861. [Google Scholar] [CrossRef]
- Reis, H.B.D.; Carvalho, M.E.; Espigolan, R.; Poleti, M.D.; Ambrizi, D.R.; Berton, M.P.; Ferraz, J.B.S.; De Mattos Oliveira, E.C.; Eler, J.P. Genome-Wide Association (GWAS) Applied to Carcass and Meat Traits of Nellore Cattle. Metabolites 2023, 14, 6. [Google Scholar] [CrossRef] [PubMed]
- Oliveira Júnior, G.A.; Santos, D.J.A.; Cesar, A.S.M.; Boison, S.A.; Ventura, R.V.; Perez, B.C.; Garcia, J.F.; Ferraz, J.B.S.; Garrick, D.J. Fine mapping of genomic regions associated with female fertility in Nellore beef cattle based on sequence variants from segregating sires. J. Anim. Sci. Biotechnol. 2019, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Börnigen, D.; Tranchevent, L.C.; Bonachela-Capdevila, F.; Devriendt, K.; De Moor, B.; De Causmaecker, P.; Moreau, Y. An unbiased evaluation of gene prioritization tools. Bioinformatics 2012, 28, 3081–3088. [Google Scholar] [CrossRef]
- Azadifar, S.; Ahmadi, A. A novel candidate disease gene prioritization method using deep graph convolutional networks and semi-supervised learning. BMC Bioinform. 2022, 23, 422. [Google Scholar] [CrossRef]
- Fonseca, P.A.D.S.; Dos Santos, F.C.; Lam, S.; Suárez-Vega, A.; Miglior, F.; Schenkel, F.S.; Diniz, L.D.A.F.; Id-Lahoucine, S.; Carvalho, M.R.S.; Cánovas, A. Genetic mechanisms underlying spermatic and testicular traits within and among cattle breeds: Systematic review and prioritization of GWAS results. J. Anim. Sci. 2018, 96, 4978–4999. [Google Scholar] [CrossRef]
- Narayana, S.G.; de Jong, E.; Schenkel, F.S.; Fonseca, P.A.S.; Chud, T.C.S.; Powell, D.; Wachoski-Dark, G.; Ronksley, P.E.; Miglior, F.; Orsel, K.; et al. Underlying genetic architecture of resistance to mastitis in dairy cattle: A systematic review and gene prioritization analysis of genome-wide association studies. J. Dairy Sci. 2023, 106, 323–351. [Google Scholar] [CrossRef]
- Silva, T.D.L.; Gondro, C.; Fonseca, P.A.D.S.; Da Silva, D.A.; Vargas, G.; Neves, H.H.D.R.; Filho, I.C.; Teixeira, C.D.S.; Albuquerque, L.G.D.; Carvalheiro, R. Testicular hypoplasia in Nellore Cattle: Genetic analysis and functional analysis of genome-wide association study results. J. Anim. Breed. Genet. 2023, 140, 185–197. [Google Scholar] [CrossRef]
- Silva, T.D.L.; Gondro, C.; Fonseca, P.A.D.S.; Silva, D.A.D.; Vargas, G.; Neves, H.H.D.R.; Carvalho Filho, I.; Teixeira, C.D.S.; Albuquerque, L.G.D.; Carvalheiro, R. Feet and legs malformation in Nellore cattle: Genetic analysis and prioritization of GWAS results. Front. Genet. 2023, 14, 1118308. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, n71, 372. [Google Scholar] [CrossRef]
- Sarkis-Onofre, R.; Catalá-López, F.; Aromataris, E.; Lockwood, C. How to properly use the PRISMA Statement. Syst. Rev. 2021, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the future: Meeting new challenges and providing updated services. Nucleic Acids Res. 2022, 50, D956–D961. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, P.A.S.; Suárez-Vega, A.; Marras, G.; Cánovas, Á. GALLO: An R package for genomic annotation and integration of multiple data sources in livestock for positional candidate loci. GigaScience 2020, 9, giaa149. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016; Available online: https://www.R-project.org/ (accessed on 12 January 2024).
- Nicolazzi, E.L.; Caprera, A.; Nazzicari, N.; Cozzi, P.; Strozzi, F.; Lawley, C.; Pirani, A.; Soans, C.; Brew, F.; Jorjani, H.; et al. SNPchiMp v.3: Integrating and standardizing single nucleotide polymorphism data for livestock species. BMC Genom. 2015, 16, 283. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.J.; Amode, M.R.; Aneja, A.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J.; et al. Ensembl 2023. Nucleic Acids Res. 2023, 51, D933–D941. [Google Scholar] [CrossRef]
- Shamimuzzaman, M.; Le Tourneau, J.J.; Unni, D.R.; Diesh, C.M.; Triant, D.A.; Walsh, A.T.; Tayal, A.; Conant, G.C.; Hagen, D.E.; Elsik, C.G. Bovine Genome Database: New annotation tools for a new reference genome. Nucleic Acids Res. 2020, 48, D676–D681. [Google Scholar] [CrossRef]
- Raney, B.J.; Barber, G.P.; Benet-Pagès, A.; Casper, J.; Clawson, H.; Cline, M.S.; Diekhans, M.; Fischer, C.; Navarro Gonzalez, J.; Hickey, G.; et al. The UCSC Genome Browser database: 2024 update. Nucleic Acids Res. 2024, 52, D1082–D1088. [Google Scholar] [CrossRef]
- Nelson, S.J.; Schopen, M.; Savage, A.G.; Schulman, J.L.; Arluk, N. The MeSH translation maintenance system: Structure, interface design, and implementation. Stud. Health Technol. Inform. 2004, 107 Pt 1, 67–69. [Google Scholar]
- Adams, W.T.; Skopek, T.R. Statistical test for the comparison of samples from mutational spectra. J. Mol. Biol. 1987, 194, 391–396. [Google Scholar] [CrossRef]
- Morota, G.; Peñagaricano, F.; Petersen, J.L.; Ciobanu, D.C.; Tsuyuzaki, K.; Nikaido, I. An application of MeSH enrichment analysis in livestock. Anim. Genet. 2015, 46, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Tsuyuzaki, K.; Morota, G.; Ishii, M.; Nakazato, T.; Miyazaki, S.; Nikaido, I. MeSH ORA framework: R/Bioconductor packages to support MeSH over-representation analysis. BMC Bioinform. 2015, 16, 45. [Google Scholar] [CrossRef] [PubMed]
- Yu, G. Using meshes for MeSH term enrichment and semantic analyses. Bioinformatics 2018, 34, 3766–3767. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [PubMed]
- Guney, E.; Garcia-Garcia, J.; Oliva, B. GUILDify: A web server for phenotypic characterization of genes through biological data integration and network-based prioritization algorithms. Bioinformatics 2014, 30, 1789–1790. [Google Scholar] [CrossRef]
- Aguirre-Plans, J.; Piñero, J.; Sanz, F.; Furlong, L.I.; Fernandez-Fuentes, N.; Oliva, B.; Guney, E. GUILDify v2.0: A Tool to Identify Molecular Networks Underlying Human Diseases, Their Comorbidities and Their Druggable Targets. J. Mol. Biol. 2019, 431, 2477–2484. [Google Scholar] [CrossRef]
- Kominakis, A.; Hager-Theodorides, A.L.; Zoidis, E.; Saridaki, A.; Antonakos, G.; Tsiamis, G. Combined GWAS and ‘guilt by association’-based prioritization analysis identifies functional candidate genes for body size in sheep. Genet. Sel. Evol. 2017, 49, 41. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Sweett, H.; Fonseca, P.A.S.; Suárez-Vega, A. Genome-wide association study to identify genomic regions and positional candidate genes associated with male fertility in beef cattle. Sci Rep 2020, 10, 20102. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Morris, Q. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38 (Suppl. S2), W214–W220. [Google Scholar] [CrossRef]
- Soares, R.A.N.; Vargas, G.; Muniz, M.M.M.; Soares, M.A.M.; Cánovas, A.; Schenkel, F.; Squires, E.J. Differential gene expression in dairy cows under negative energy balance and ketosis: A systematic review and meta-analysis. J. Dairy Sci. 2021, 104, 602–615. [Google Scholar] [CrossRef]
- Oshimori, N.; Li, X.; Ohsugi, M.; Yamamoto, T. Cep72 regulates the localization of key centrosomal proteins and proper bipolar spindle formation. EMBO J. 2009, 28, 2066–2076. [Google Scholar] [CrossRef] [PubMed]
- Alliance of Genome Resources. Tubulin-γ Complex-Associated Protein 2 (TUBGCP2). 2022. Available online: www.alliancegenome.org/gene/HGNC:18599 (accessed on 27 July 2024).
- Alliance of Genome Resources. Espin Like (ESPNL). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:27937 (accessed on 27 July 2024).
- Alliance of Genome Resources. Solute Carrier Family 27 Member 1 (SLC27A1). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:27937 (accessed on 27 July 2024).
- Alliance of Genome Resources. Acyl-CoA Synthetase Medium Chain Family Member 2A (ACSM2A). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:32017 (accessed on 27 July 2024).
- Oshimori, N.; Ohsugi, M.; Yamamoto, T. The Plk1 target Kizuna stabilizes mitotic centrosomes to ensure spindle bipolarity. Nat. Cell Biol. 2006, 8, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, J.; Wu, Y.; Zhao, X.; Hao, Y.; Wang, X.; Xue, C.; Wang, Y.; Zhang, R.; Zhang, X. Long Noncoding RNA SERTAD2-3 Inhibits Osteosarcoma Proliferation and Migration by Competitively Binding miR-29c. Genet. Test. Mol. Biomark. 2020, 24, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Darwish, H. Genomic and Functional Studies of SERTAD3, an Oncogenic Protein of the SERTAD Family of Transcription Factors. Master’s Thesis, McGill University, Montreal, QC, Canada, 2006. Available online: https://escholarship.mcgill.ca/concern/theses/41687n49h (accessed on 27 July 2024).
- Fu, X.; Yucer, N.; Liu, S.; Li, M.; Yi, P.; Mu, J.J.; Yang, T.; Chu, J.; Jung, S.Y.; O’Malley, B.W.; et al. RFWD3-Mdm2 ubiquitin ligase complex positively regulates p53 stability in response to DNA damage. Proc. Natl. Acad. Sci. USA 2010, 107, 4579–4584. [Google Scholar] [CrossRef]
- Lamsam-Casalotti, S.; Onoda, M.; Papadopoulos, V.; Dym, M. Developmental expression of GTP-binding proteins in rat testes. J. Reprod. Fertil. 1993, 99, 487–495. [Google Scholar] [CrossRef]
- Alliance of Genome Resources. Acyl-CoA Synthetase Medium Chain Family Member 4 (ACSM4). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:32016 (accessed on 27 July 2024).
- Manna, P.R.; Stetson, C.L.; Slominski, A.T.; Pruitt, K. Role of the steroidogenic acute regulatory protein in health and disease. Endocrine 2016, 51, 7–21. [Google Scholar] [CrossRef]
- Errico, A.; Vinco, S.; Ambrosini, G.; Dalla Pozza, E.; Marroncelli, N.; Zampieri, N.; Dando, I. Mitochondrial Dynamics as Potential Modulators of Hormonal Therapy Effectiveness in Males. Biology 2023, 12, 547. [Google Scholar] [CrossRef]
- Alliance of Genome Resources. AlkB Homolog 7 (ALKBH7). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:21306 (accessed on 27 July 2024).
- Alliance of Genome Resources. Codanin 1 (CDAN1). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:1713 (accessed on 27 July 2024).
- Alliance of Genome Resources. Dual Specificity Tyrosine Phosphorylation Regulated Kinase 2 (DYRK2). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:3093 (accessed on 27 July 2024).
- Alliance of Genome Resources. Heat Shock Protein Family A (Hsp70) Member 1A (HSPA1A). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:5232 (accessed on 27 July 2024).
- Alliance of Genome Resources. Synaptosome Associated Protein 47 (SNAP47). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:30669 (accessed on 27 July 2024).
- Alliance of Genome Resources. ORC Ubiquitin Ligase 1(OBI1). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:20308 (accessed on 27 July 2024).
- Nassar, J. Studying the function(s) of OBI1, a novel E3 ubiquitin ligase, involved in DNA replication [Doctoral thesis]. HAL Open Arch. 2019. Available online: https://theses.hal.science/tel-03403352.
- Alliance of Genome Resources. Transmembrane Protein 120A (TMEM120A). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:21697 (accessed on 27 July 2024).
- Batrakou, D.G.; De Las Heras, J.I.; Czapiewski, R.; Mouras, R.; Schirmer, E.C. TMEM120A and B: Nuclear Envelope Transmembrane Proteins Important for Adipocyte Differentiation. PLoS ONE 2015, 10, e0127712. [Google Scholar] [CrossRef]
- Alliance of Genome Resources. Dual Specificity Phosphatase 29 (DUSP29). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:23481 (accessed on 27 July 2024).
- Cooper, L.M.; West, R.C.; Hayes, C.S.; Waddell, D.S. Dual-specificity phosphatase 29 is induced during neurogenic skeletal muscle atrophy and attenuates glucocorticoid receptor activity in muscle cell culture. Am. J. Physiol. Cell Physiol. 2020, 319, C441–C454. [Google Scholar] [CrossRef]
- Alliance of Genome Resources. Josephin Domain Containing 1 (JOSD1). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:28953 (accessed on 27 July 2024).
- Seki, T.; Gong, L.; Williams, A.J.; Sakai, N.; Todi, S.V.; Paulson, H.L. JosD1, a membrane-targeted deubiquitinating enzyme, is activated by ubiquitination and regulates membrane dynamics, cell motility, and endocytosis. J. Biol. Chem. 2013, 288, 17145–17155. [Google Scholar] [CrossRef] [PubMed]
- GeneCards. LOXL1 Antisense RNA 1 (LOXL1-AS1). Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=LOXL1-AS1 (accessed on 27 July 2024).
- Alliance of Genome Resources. Calcium Regulated Heat Stable Protein 1 (CARHSP1). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:17150 (accessed on 27 July 2024).
- Kociucka, B.; Stachecka, J.; Szydlowski, M.; Szczerbal, I. Rapid Communication: The correlation between histone modifications and expression of key genes involved in accumulation of adipose tissue in the pig. J. Anim. Sci. 2017, 95, 4514–4519. [Google Scholar] [CrossRef]
- Alliance of Genome Resources. Translocase of Inner Mitochondrial Membrane 8A (TIMM8A). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:11817 (accessed on 27 July 2024).
- Alliance of Genome Resources. Terminal Nucleotidyltransferase 4A (TENT4A). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:16705 (accessed on 27 July 2024).
- Alliance of Genome Resources. Ankyrin Repeat Domain 34B (ANKRD34B). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:33736 (accessed on 27 July 2024).
- Tartaglia, G.G. The Grand Challenge of Characterizing Ribonucleoprotein Networks. Front. Mol. Biosci. 2016, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Patterns and evolutionary consequences of pleiotropy. Annu. Rev. Ecol. Evol. Syst. 2023, 54, 1–19. [Google Scholar] [CrossRef]
- Alliance of Genome Resources. Ring Finger and WD Repeat Domain 3 (RFWD3). 2022. Available online: https://www.alliancegenome.org/gene/HGNC:25539 (accessed on 27 July 2024).
- Ménézo, Y.; Dale, B.; Cohen, M. DNA damage and repair in human oocytes and embryos: A review. Zygote 2010, 18, 357–365. [Google Scholar] [CrossRef]
- Inano, S.; Sato, K.; Katsuki, Y.; Kobayashi, W.; Tanaka, H.; Nakajima, K.; Nakada, S.; Miyoshi, H.; Knies, K.; Takaori-Kondo, A.; et al. RFWD3-Mediated Ubiquitination Promotes Timely Removal of Both RPA and RAD51 from DNA Damage Sites to Facilitate Homologous Recombination. Mol. Cell 2017, 66, 622–634.e8. [Google Scholar] [CrossRef] [PubMed]
- Alvarenga, A.B.; Oliveira, H.R.; Chen, S.Y.; Miller, S.P.; Marchant-Forde, J.N.; Grigoletto, L.; Brito, L.F. A Systematic Review of Genomic Regions and Candidate Genes Underlying Behavioral Traits in Farmed Mammals and Their Link with Human Disorders. Anim. Open Access J. 2021, 11, 715. [Google Scholar] [CrossRef]
- Bovine Genome Sequencing and Analysis Consortium; Elsik, C.G.; Tellam, R.L.; Worley, K.C.; Gibbs, R.A.; Muzny, D.M.; Weinstock, G.M.; Adelson, D.L.; Eichler, E.E.; Elnitski, L.; et al. The genome sequence of taurine cattle: A window to ruminant biology and evolution. Science 2009, 324, 522–528. [Google Scholar] [CrossRef]
- Weitzman, J.B. Comparing cows with humans. Genome Biol. 2000, 1, spotlight-20001005-04. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 Trait Groups | Studies | Genomic Regions | Candidate Genes |
|---|---|---|---|
| Reproduction | 23 | 605 | 8569 |
| Sexual precocity | 19 | 387 | 5412 |
| Carcass | 14 | 607 | 11,195 |
| Meat quality | 8 | 294 | 6646 |
| Growth | 12 | 360 | 5239 |
| Efficiency | 7 | 217 | 3319 |
| Morphological | 5 | 189 | 3483 |
| Visual scores | 3 | 132 | 2114 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogunbawo, A.R.; Mulim, H.A.; Campos, G.S.; Oliveira, H.R. Genetic Foundations of Nellore Traits: A Gene Prioritization and Functional Analyses of Genome-Wide Association Study Results. Genes 2024, 15, 1131. https://doi.org/10.3390/genes15091131
Ogunbawo AR, Mulim HA, Campos GS, Oliveira HR. Genetic Foundations of Nellore Traits: A Gene Prioritization and Functional Analyses of Genome-Wide Association Study Results. Genes. 2024; 15(9):1131. https://doi.org/10.3390/genes15091131
Chicago/Turabian StyleOgunbawo, Adebisi R., Henrique A. Mulim, Gabriel S. Campos, and Hinayah R. Oliveira. 2024. "Genetic Foundations of Nellore Traits: A Gene Prioritization and Functional Analyses of Genome-Wide Association Study Results" Genes 15, no. 9: 1131. https://doi.org/10.3390/genes15091131
APA StyleOgunbawo, A. R., Mulim, H. A., Campos, G. S., & Oliveira, H. R. (2024). Genetic Foundations of Nellore Traits: A Gene Prioritization and Functional Analyses of Genome-Wide Association Study Results. Genes, 15(9), 1131. https://doi.org/10.3390/genes15091131