
Identifying Candidate Genes Related to Soybean (Glycine max) Seed Coat Color via RNA-Seq and Coexpression Network Analysis
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Fixation and Staining
2.3. RNA-Seq Sequencing
2.4. Identification of Differentially Expressed Genes
2.5. Construction of the Coexpression Network
2.6. Quantitative Real-Time PCR
3. Results
3.1. Distribution of Anthocyanins in Different Soybean Seed Coat Colors
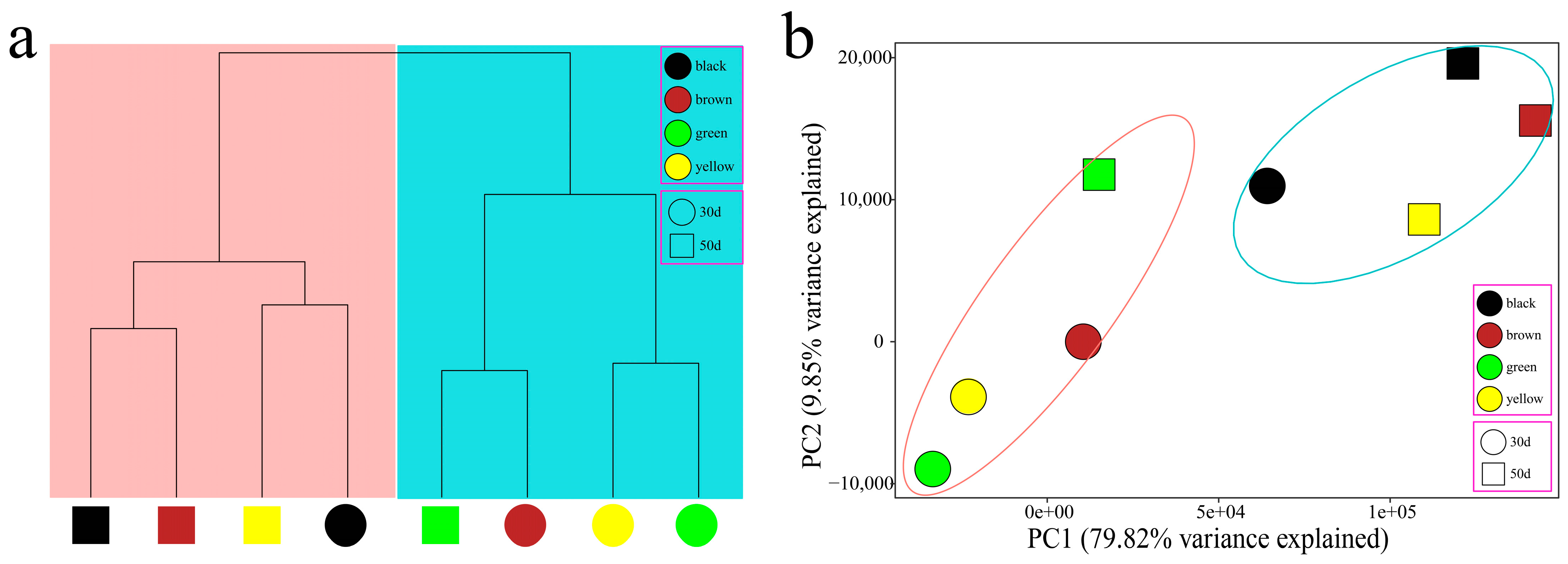
3.2. Overall Analysis of RNA-Seq Data
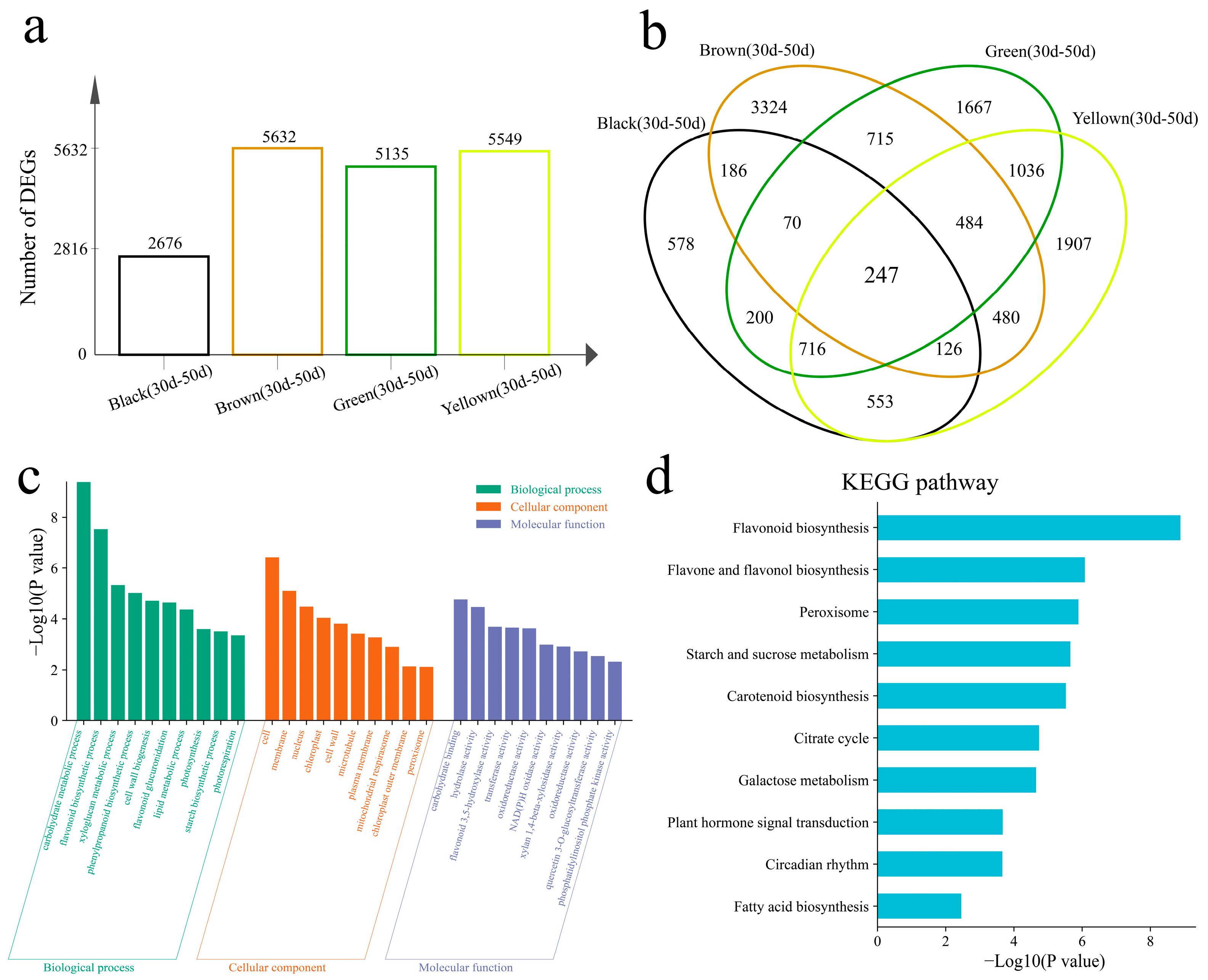
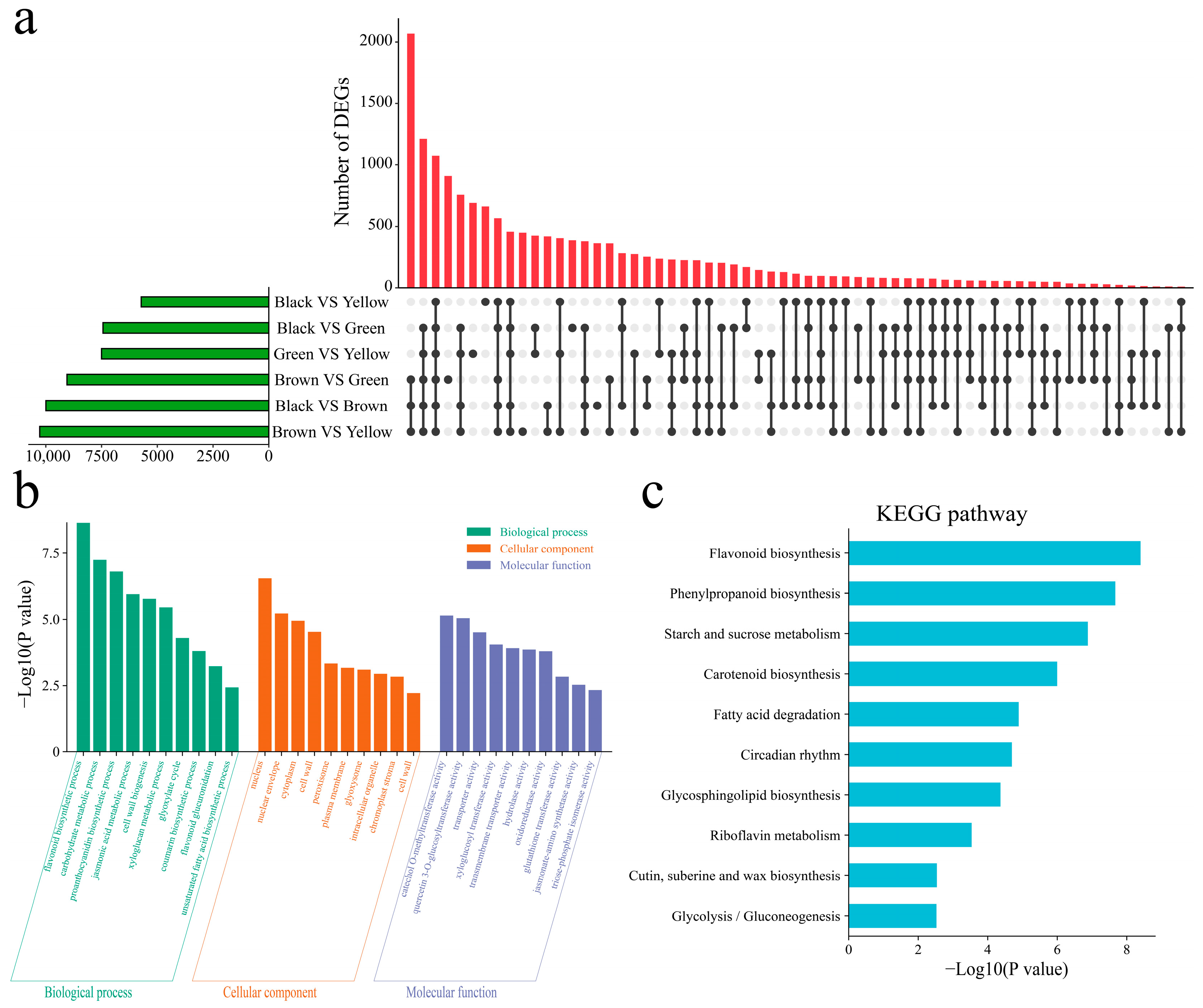
3.3. Difference Analysis
3.4. Analysis of Differentially Expressed Transcription Factors
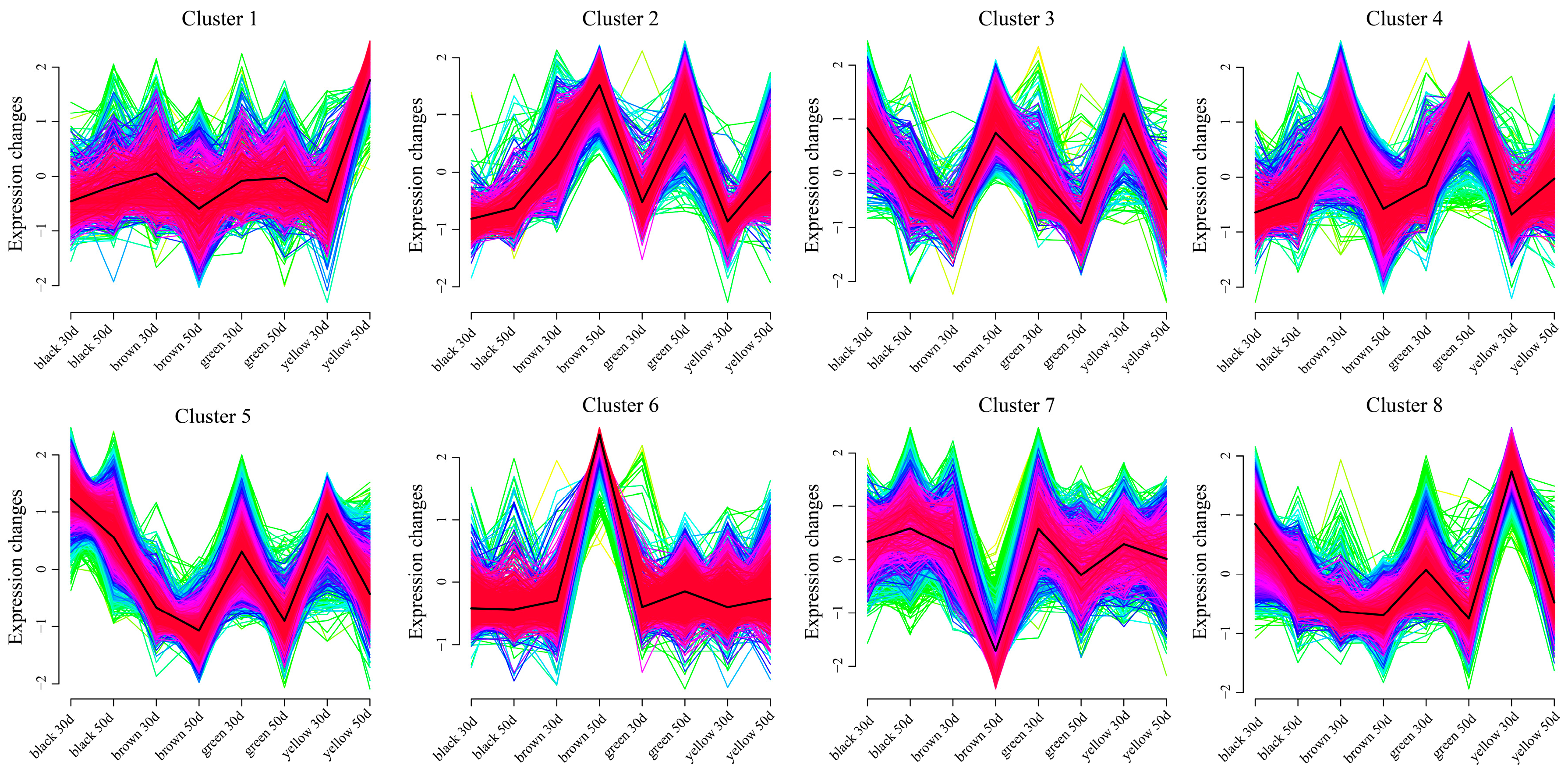
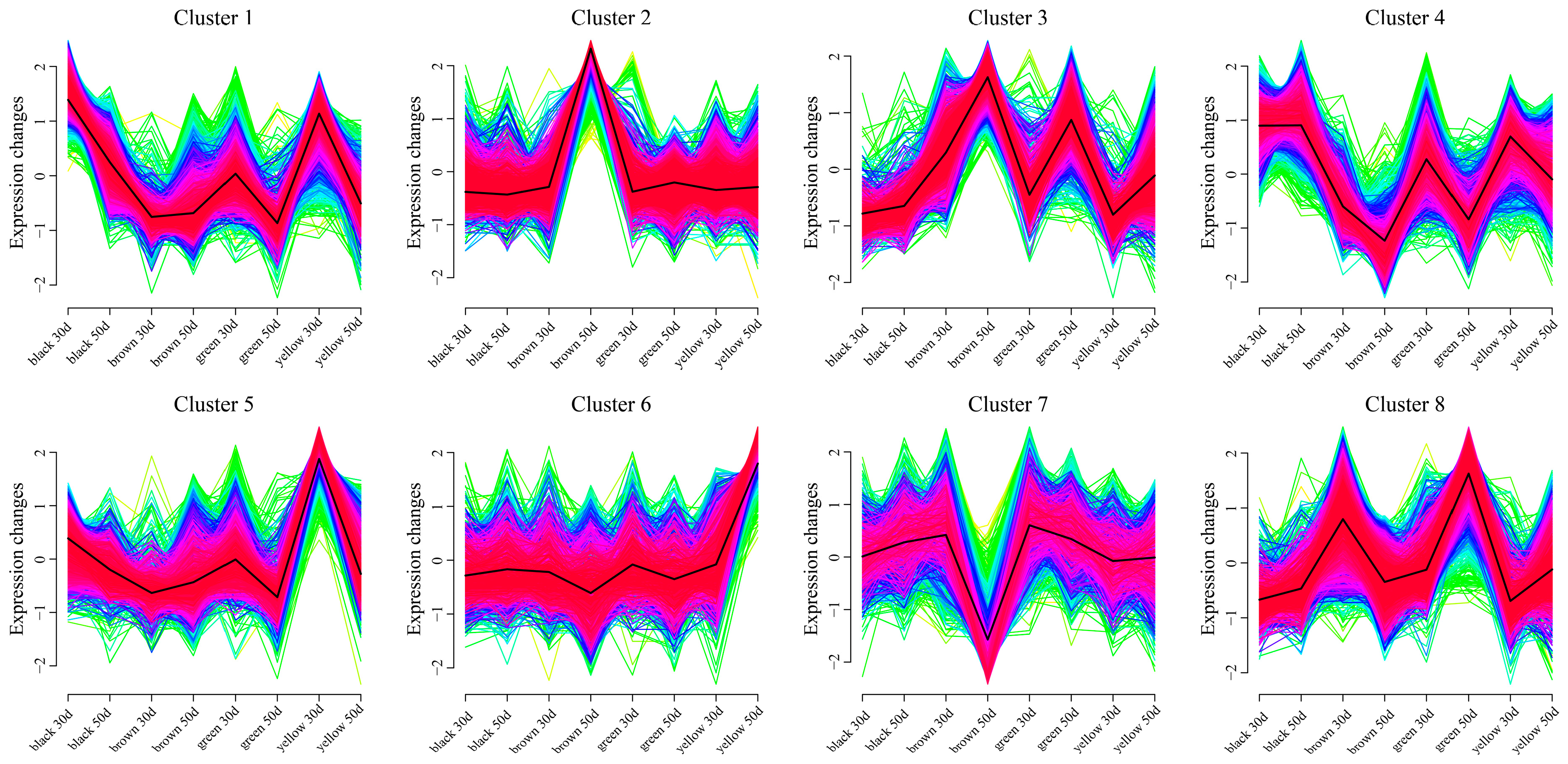
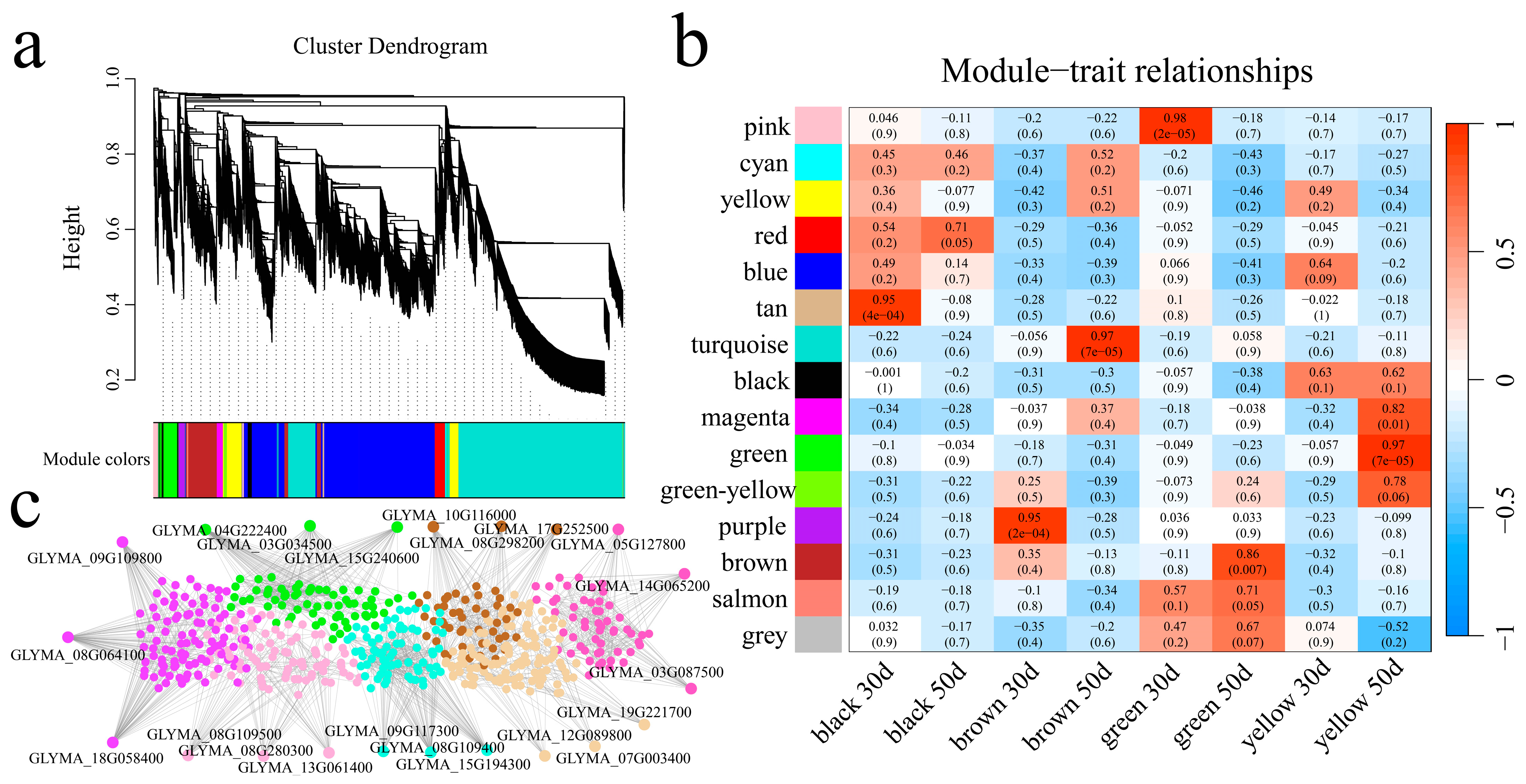
3.5. Weighted Gene Coexpression Network Construction and Candidate Gene Mining
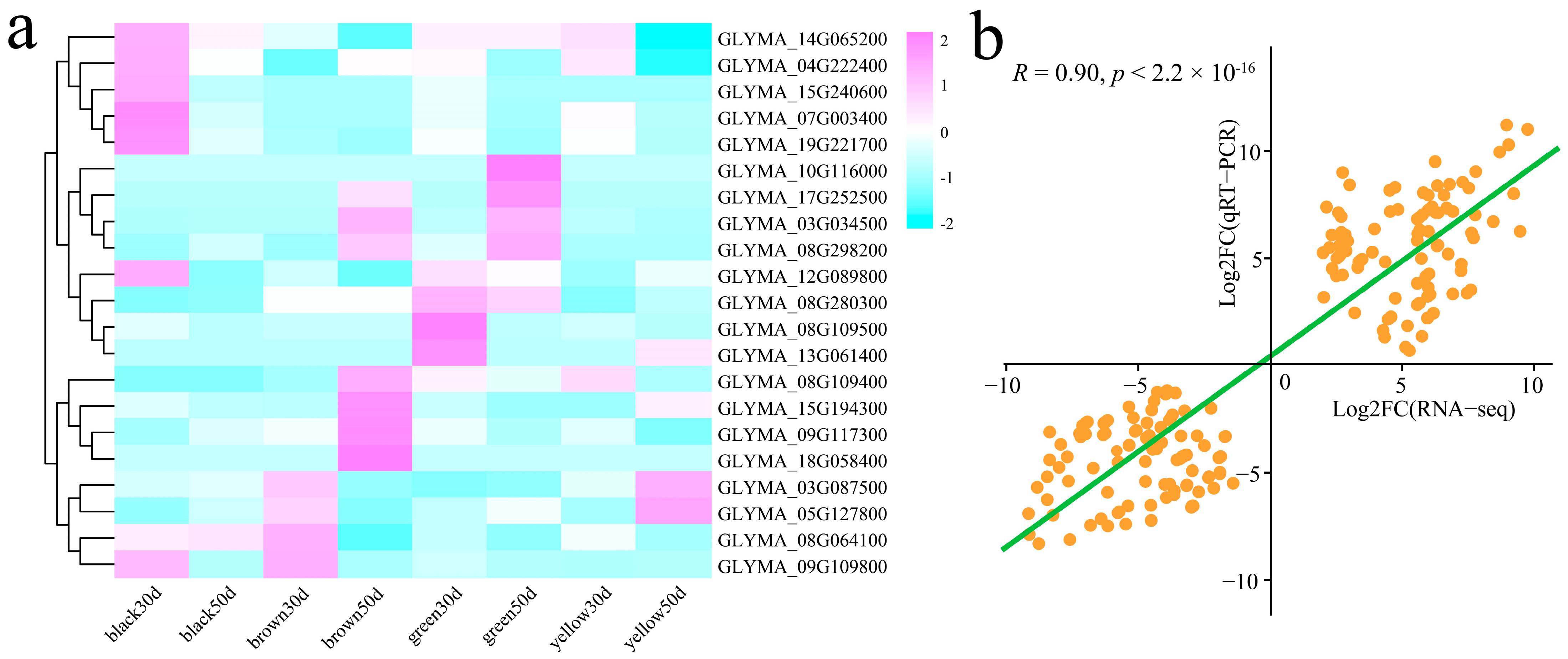
3.6. qRT–PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Duan, Z.; Li, Q.; Wang, H.; He, X.; Zhang, M. Genetic regulatory networks of soybean seed size, oil and protein contents. Front. Plant Sci. 2023, 14, 1160418. [Google Scholar] [CrossRef] [PubMed]
- Kudełka, W.; Kowalska, M.; Popis, M. Quality of soybean products in terms of essential amino acids composition. Molecule 2021, 26, 5071. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Jeong, N.; Moon, J.K.; Lee, Y.H.; Lee, S.H.; Kim, H.M.; Hwang, C.H.; Back, K.; Palmer, R.G.; Jeong, S.C. Genetic analysis of genes controlling natural variation of seed coat and flower colors in soybean. J. Hered. 2010, 101, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Kafer, J.M.; Molinari, M.D.C.; Henning, F.A.; Koltun, A.; Marques, V.V.; Marin, S.R.R.; Nepomuceno, A.L.; Mertz-Henning, L.M. Transcriptional profile of soybean seeds with contrasting seed coat color. Plants 2023, 12, 1555. [Google Scholar] [CrossRef]
- Kovinich, N.; Saleem, A.; Arnason, J.T.; Miki, B. Combined analysis of transcriptome and metabolite data reveals extensive differences between black and brown nearly-isogenic soybean (Glycine max) seed coats enabling the identification of pigment isogenes. BMC Genom. 2011, 12, 381. [Google Scholar] [CrossRef] [PubMed]
- Shee, C.N.; Lemenager, R.P.; Schoonmaker, J.P. Feeding dried distillers grains with solubles to lactating beef cows: Impact of excess protein and fat on post-weaning progeny growth, glucose tolerance and carcass traits. Animal 2018, 12, 750–756. [Google Scholar] [CrossRef]
- Dixon, R.A.; Sumner, L.W. Legume natural products: Understanding and manipulating complex pathways for human and animal health. Plant Physiol. 2003, 131, 878–885. [Google Scholar] [CrossRef]
- Owen, F.V. Inheritance Studies in Soybeans. III. Seed-Coat Color and Summary of All Other Mendelian Characters Thus Far Reported. Genetics 1928, 13, 50–79. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.G.; Pfeiffer, T.W.; Buss, G.R.; Kilen, T.C.; Boerma, H.R.; Specht, J.E. Soybeans: Improvement, production, and uses. Agronomy 2004, 137–233. [Google Scholar]
- Todd, J.J.; Vodkin, L.O. Pigmented Soybean (Glycine max) seed coats accumulate proanthocyanidins during development. Plant Physiol. 1993, 102, 663–670. [Google Scholar] [CrossRef]
- Buzzell, R.I.; Buttery, B.R.; MacTavish, D.C. Biochemical genetics of black pigmentation of soybean seed. J. Hered. 1987, 78, 53–54. [Google Scholar] [CrossRef]
- He, J.; Giusti, M.M. Anthocyanins: Natural colorants with health-promoting properties. Annu. Rev. Food Sci. Technol. 2010, 1, 163–187. [Google Scholar] [CrossRef]
- Kovinich, N.; Arnason, J.T.; De Luca, V.; Miki, B. Coloring soybeans with anthocyanins? Biol. Act. Phytochem. 2011, 41, 47–57. [Google Scholar] [CrossRef]
- Clough, S.J.; Tuteja, J.H.; Li, M.; Marek, L.F.; Shoemaker, R.C.; Vodkin, L.O. Features of a 103-kb gene-rich region in soybean include an inverted perfect repeat cluster of CHS genes comprising the I locus. Genome 2004, 47, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Montazerinezhad, S.; Solouki, M.; Emamjomeh, A.; Kavousi, K.; Taheri, A.; Shiri, Y. Transcriptomic analysis of alternative splicing events for different stages of growth and development in Sistan Yaghooti grape clusters. Gene 2024, 896, 148030. [Google Scholar] [CrossRef] [PubMed]
- Pires, R.C.; Ferro, A.; Capote, T.; Usié, A.; Correia, B.; Pinto, G.; Menéndez, E.; Marum, L. Laser Microdissection of Woody and Suberized Plant Tissues for RNA-Seq Analysis. Mol. Biotechnol. 2023, 65, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Dai, B.; Luo, Y.; Ding, Y. Integrated analysis of multiple metabolome and transcriptome revealed the accumulation of flavonoids and associated molecular regulation mechanisms in Rubus chingii Hu at different developmental stages. Plant Physiol. Biochem. 2023, 204, 108085. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, C.; Li, Y.; Wang, X.; Pan, X.; Wang, F.; Zhang, Q. Developmental dynamic transcriptome and systematic analysis reveal the major genes underlying isoflavone accumulation in soybean. Front. Plant Sci. 2023, 14, 1014349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yue, Y.; Hu, M.; Yi, F.; Chen, J.; Lai, J.; Xin, B. Dynamic transcriptome landscape of maize pericarp development. Plant J. 2024, 117, 1574–1591. [Google Scholar] [CrossRef] [PubMed]
- Ohm, H.; Saripella, G.V.; Hofvander, P.; Grimberg, Å. Spatio-temporal transcriptome and storage compound profiles of developing faba bean (Vicia faba) seed tissues. Front. Plant Sci. 2024, 15, 1284997. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, Y.; Hu, M.; Zhao, Y.; Liu, B.; Wang, C.; Zhang, M.; Zhang, L.; Yang, X.; Mu, G. Multi-Omics and miRNA interaction joint analysis highlight new insights into anthocyanin biosynthesis in peanuts (Arachis hypogaea L.). Front. Plant Sci. 2022, 13, 818345. [Google Scholar] [CrossRef]
- Lyu, X.; Li, Y.H.; Li, Y.; Li, D.; Han, C.; Hong, H.; Tian, Y.; Han, L.; Liu, B.; Qiu, L.J. The domestication-associated L1 gene encodes a eucomic acid synthase pleiotropically modulating pod pigmentation and shattering in soybean. Mol. Plant 2023, 16, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Yang, J.; Li, P.; Li, Y.; Xiao, Q. GelFAP v2.0: An improved platform for Gene functional analysis in Gastrodia elata. BMC Genom. 2023, 24, 164. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Abeynayake, S.W.; Panter, S.; Mouradov, A.; Spangenberg, G. A high-resolution method for the localization of proanthocyanidins in plant tissues. Plant Methods 2011, 7, 13. [Google Scholar] [CrossRef]
- Lim, Y.J.; Kwon, S.J.; Qu, S.; Kim, D.G.; Eom, S.H. Antioxidant Contributors in Seed, Seed Coat, and Cotyledon of γ-ray-Induced Soybean Mutant Lines with Different Seed Coat Colors. Antioxidants 2021, 10, 353. [Google Scholar] [CrossRef] [PubMed]
- Gebregziabher, B.S.; Zhang, S.; Ghosh, S.; Shaibu, A.S.; Azam, M.; Abdelghany, A.M.; Qi, J.; Agyenim-Boateng, K.G.; Htway, H.T.P.; Feng, Y.; et al. Origin, Maturity Group and Seed Coat Color Influence Carotenoid and Chlorophyll Concentrations in Soybean Seeds. Plants 2022, 11, 848. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, S.; Funatsuki, H.; Kasai, A.; Kurauchi, T.; Yamaguchi, N.; Takeuchi, T.; Yamazaki, H.; Kurosaki, H.; Shirai, S.; Miyoshi, T.; et al. Variation of GmIRCHS (Glycine max inverted-repeat CHS pseudogene) is related to tolerance of low temperature-induced seed coat discoloration in yellow soybean. Theor. Appl. Genet. 2011, 122, 633–642. [Google Scholar] [CrossRef]
- Pal, L.; Dwivedi, V.; Gupta, S.K.; Saxena, S.; Pandey, A.; Chattopadhyay, D. Biochemical analysis of anthocyanin and proanthocyanidin and their regulation in determining chickpea flower and seed coat colour. J. Exp. Bot. 2023, 74, 130–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Chen, H.; Dai, G.; Zhang, H.; Liu, Y.; Shen, W.; Zhu, B.; Cui, C.; Tan, C. Genome-wide identification and expression analysis of the anthocyanin-related genes during seed coat development in six Brassica species. BMC Genom. 2023, 24, 103. [Google Scholar] [CrossRef] [PubMed]
- Sunil, L.; Shetty, N.P. Biosynthesis and regulation of anthocyanin pathway genes. Appl. Microbiol. Biotechnol. 2022, 106, 1783–1798. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, J.H.; Zabala, G.; Varala, K.; Hudson, M.; Vodkin, L.O. Endogenous, tissue-specific short interfering RNAs silence the chalcone synthase gene family in Glycine max seed coats. Plant Cell 2009, 21, 3063–3077. [Google Scholar] [CrossRef]
- Zabala, G.; Vodkin, L.O. Methylation affects transposition and splicing of a large CACTA transposon from a MYB transcription factor regulating anthocyanin synthase genes in soybean seed coats. PLoS ONE 2014, 9, e111959. [Google Scholar] [CrossRef]
- Bao, X.; Zong, Y.; Hu, N.; Li, S.; Liu, B.; Wang, H. Functional R2R3-MYB transcription factor NsMYB1, regulating anthocyanin biosynthesis, was relative to the fruit color differentiation in Nitraria sibirica Pall. BMC Plant Biol. 2022, 22, 186. [Google Scholar] [CrossRef]
- Lu, N.; Rao, X.; Li, Y.; Jun, J.H.; Dixon, R.A. Dissecting the transcriptional regulation of proanthocyanidin and anthocyanin biosynthesis in soybean (Glycine max). Plant Biotechnol. J. 2021, 19, 1429–1442. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthy, J.; Park, G.T.; Chang, J.H.; Lee, J.D.; Kim, J.H.; Seo, H.S.; Chung, G.; Song, J.T. Identification and molecular analysis of four new alleles at the W1 locus associated with flower color in soybean. PLoS ONE 2016, 11, e0159865. [Google Scholar] [CrossRef]
- Yuan, Z.; Dong, F.; Pang, Z.; Fallah, N.; Zhou, Y.; Li, Z.; Hu, C. Integrated metabolomics and transcriptome analyses unveil pathways involved in sugar content and rind color of two sugarcane varieties. Front. Plant Sci. 2022, 13, 921536. [Google Scholar] [CrossRef]
- Das, P.K.; Geul, B.; Choi, S.B.; Yoo, S.D.; Park, Y.I. Photosynthesis-dependent anthocyanin pigmentation in Arabidopsis. Plant Signal. Behav. 2011, 6, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Rolland, F.; Baena-Gonzalez, E.; Sheen, J. Sugar sensing and signaling in plants: Conserved and novel mechanisms. Annu. Rev. Plant Biol. 2006, 57, 675–709. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.W.; Das, P.K.; Jeoung, S.C.; Song, J.Y.; Lee, H.K.; Kim, Y.K.; Kim, W.J.; Park, Y.I.; Yoo, S.D.; Choi, S.B.; et al. Ethylene suppression of sugar-induced anthocyanin pigmentation in Arabidopsis. Plant Physiol. 2010, 154, 1514–1531. [Google Scholar] [CrossRef] [PubMed]
- Batschauer, A. Photoreceptors of higher plants. Planta 1998, 206, 479–492. [Google Scholar] [CrossRef]
- Chaves, I.; Pokorny, R.; Byrdin, M.; Hoang, N.; Ritz, T.; Brettel, K.; Essen, L.O.; van der Horst, G.T.; Batschauer, A.; Ahmad, M. The cryptochromes: Blue light photoreceptors in plants and animals. Annu. Rev. Plant Biol. 2011, 62, 335–364. [Google Scholar] [CrossRef]
- Jenkins, G.I. UV and blue light signal transduction in Arabidopsis. Plant Cell Environ. 1997, 20, 773–778. [Google Scholar] [CrossRef]
- Nagano, S.; Mori, N.; Tomari, Y.; Mitsugi, N.; Deguchi, A.; Kashima, M.; Tezuka, A.; Nagano, A.J.; Usami, H.; Tanabata, T.; et al. Effect of differences in light source environment on transcriptome of leaf lettuce (Lactuca sativa L.) to optimize cultivation conditions. PLoS ONE 2022, 17, e0265994. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Le Gourrierec, J.; Baudry, A.; Huep, G.; Lanet, E.; Debeaujon, I.; Routaboul, J.M.; Alboresi, A.; Weisshaar, B.; Lepiniec, L. MYBL2 is a new regulator of flavonoid biosynthesis in Arabidopsis thaliana. Plant J. 2008, 55, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Zoratti, L.; Karppinen, K.; Luengo Escobar, A.; Häggman, H.; Jaakola, L. Light-controlled flavonoid biosynthesis in fruits. Front. Plant Sci. 2014, 5, 534. [Google Scholar] [CrossRef] [PubMed]
- Rameneni, J.J.; Choi, S.R.; Chhapekar, S.S.; Kim, M.S.; Singh, S.; Yi, S.Y.; Oh, S.H.; Kim, H.; Lee, C.Y.; Oh, M.H.; et al. Red Chinese cabbage transcriptome analysis reveals structural genes and multiple transcription factors regulating reddish purple color. Int. J. Mol. Sci. 2020, 21, 2901. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Jeong, C.Y.; Kang, G.H.; Yoo, S.D.; Hong, S.W.; Lee, H. MYBD employed by HY5 increases anthocyanin accumulation via repression of MYBL2 in Arabidopsis. Plant J. 2015, 84, 1192–1205. [Google Scholar] [CrossRef]
- Bi, M.; Liang, R.; Wang, J.; Qu, Y.; Liu, X.; Cao, Y.; He, G.; Yang, Y.; Yang, P.; Xu, L.; et al. Multifaceted roles of LhWRKY44 in promoting anthocyanin accumulation in Asiatic hybrid lilies (Lilium spp.). Hortic. Res. 2023, 10, uhad167. [Google Scholar] [CrossRef]
- Gao, R.; Han, T.; Xun, H.; Zeng, X.; Li, P.; Li, Y.; Wang, Y.; Shao, Y.; Cheng, X.; Feng, X.; et al. MYB transcription factors GmMYBA2 and GmMYBR function in a feedback loop to control pigmentation of seed coat in soybean. J. Exp. Bot. 2021, 72, 4401–4418. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | COG | GO | KEGG | KOG | Pfam | Swissprot | TrEMBL | Nr | All |
|---|---|---|---|---|---|---|---|---|---|
| Number | 17,860 | 42,447 | 11,047 | 28,613 | 43,237 | 42,272 | 55,817 | 56,835 | 56,848 |
| Gene Id | Gene Name | Functional Annotation |
|---|---|---|
| GLYMA_08G109500 | CHS | Flavonoid biosynthetic process |
| GLYMA_08G280300 | AP2/ERF | Seed development |
| GLYMA_03G034500 | NB-ARC | Defense response |
| GLYMA_03G087500 | MYB | Anthocyanin synthesis |
| GLYMA_04G222400 | CHI | Flavonoid biosynthetic process |
| GLYMA_05G127800 | Probable starch synthase 4 | Starch biosynthetic process |
| GLYMA_07G003400 | bHLH | L-serine biosynthetic process |
| GLYMA_08G064100 | L-galactose dehydrogenase | Amino acid metabolic process |
| GLYMA_08G109400 | CHS | Flavonoid biosynthetic process |
| GLYMA_08G298200 | MYB | Cell cycle |
| GLYMA_09G109800 | MADS | Floral organ development |
| GLYMA_09G117300 | SDR | Oxidoreductase activity |
| GLYMA_10G116000 | Cytochrome P450 | Secondary metabolites biosynthesis |
| GLYMA_12G089800 | Calmodulin-like | Calcium ion binding |
| GLYMA_13G061400 | UDP-glycosyltransferase | Flavonoid biosynthetic process |
| GLYMA_14G065200 | Serine/threonine-protein phosphatase | Serine/threonine phosphatase activity |
| GLYMA_15G194300 | Photosystem I reaction center subunit psaK | Photosynthesis |
| GLYMA_15G240600 | F3′H | Flavonoid biosynthetic process |
| GLYMA_17G252500 | Glucose-1-phosphate | Starch biosynthetic process |
| GLYMA_18G058400 | lipid phosphate phosphatase | Phospholipid metabolic process |
| GLYMA_19G221700 | WRKY | Fruit development |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Fu, P.; Sun, T.; Wang, Y.; Li, X.; Lan, S.; Liu, H.; Gou, Y.; Shang, Q.; Li, W. Identifying Candidate Genes Related to Soybean (Glycine max) Seed Coat Color via RNA-Seq and Coexpression Network Analysis. Genes 2025, 16, 44. https://doi.org/10.3390/genes16010044
Wang C, Fu P, Sun T, Wang Y, Li X, Lan S, Liu H, Gou Y, Shang Q, Li W. Identifying Candidate Genes Related to Soybean (Glycine max) Seed Coat Color via RNA-Seq and Coexpression Network Analysis. Genes. 2025; 16(1):44. https://doi.org/10.3390/genes16010044
Chicago/Turabian StyleWang, Cheng, Pingchun Fu, Tingting Sun, Yan Wang, Xueting Li, Shulin Lan, Hui Liu, Yongji Gou, Qiaoxia Shang, and Weiyu Li. 2025. "Identifying Candidate Genes Related to Soybean (Glycine max) Seed Coat Color via RNA-Seq and Coexpression Network Analysis" Genes 16, no. 1: 44. https://doi.org/10.3390/genes16010044
APA StyleWang, C., Fu, P., Sun, T., Wang, Y., Li, X., Lan, S., Liu, H., Gou, Y., Shang, Q., & Li, W. (2025). Identifying Candidate Genes Related to Soybean (Glycine max) Seed Coat Color via RNA-Seq and Coexpression Network Analysis. Genes, 16(1), 44. https://doi.org/10.3390/genes16010044