
UCN-Centric Prognostic Model for Predicting Overall Survival and Immune Response in Colorectal Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Data Source
2.3. Weighted Gene Co-Expression Network Analysis (WGCNA)
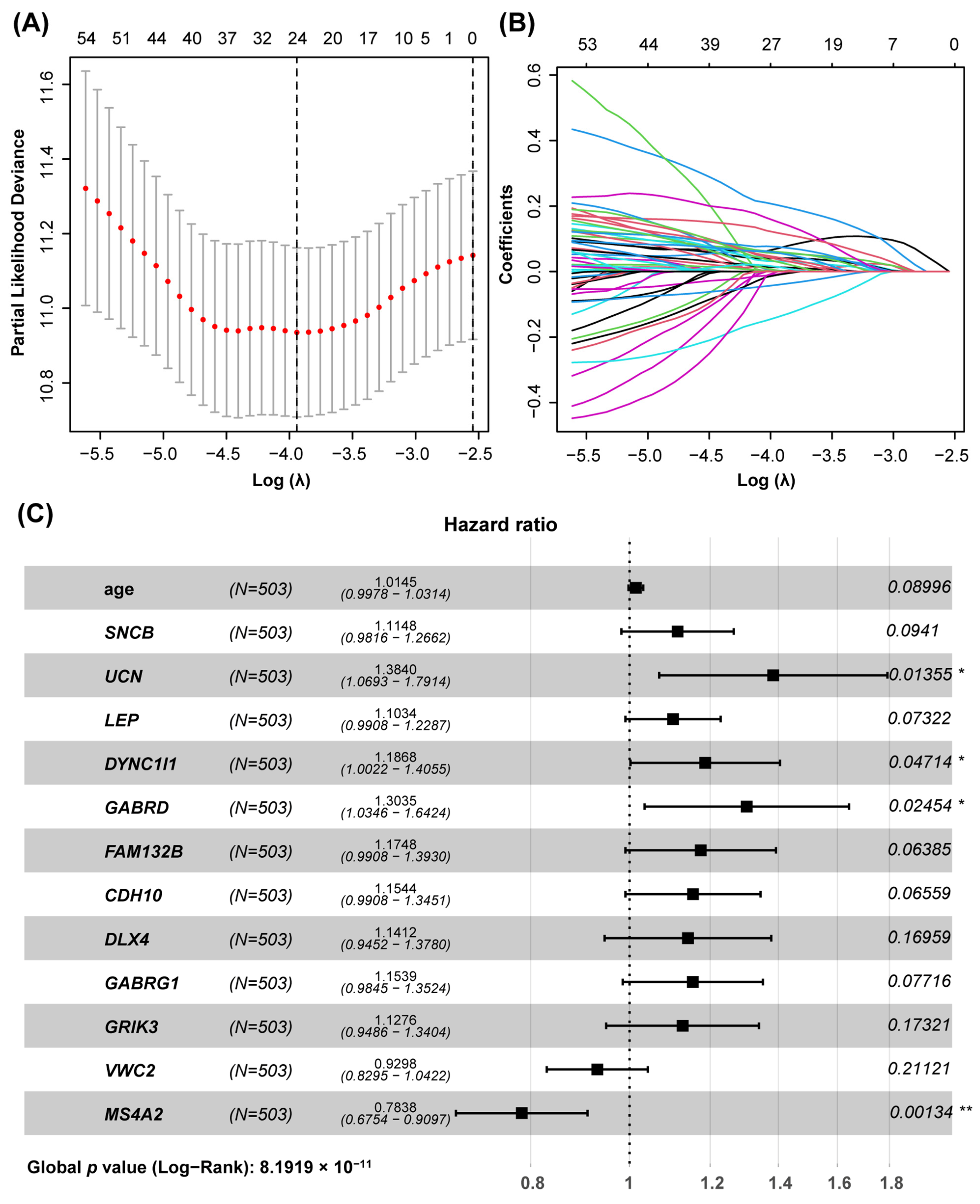
2.4. Least Absolute Shrinkage and Selection Operator (LASSO)
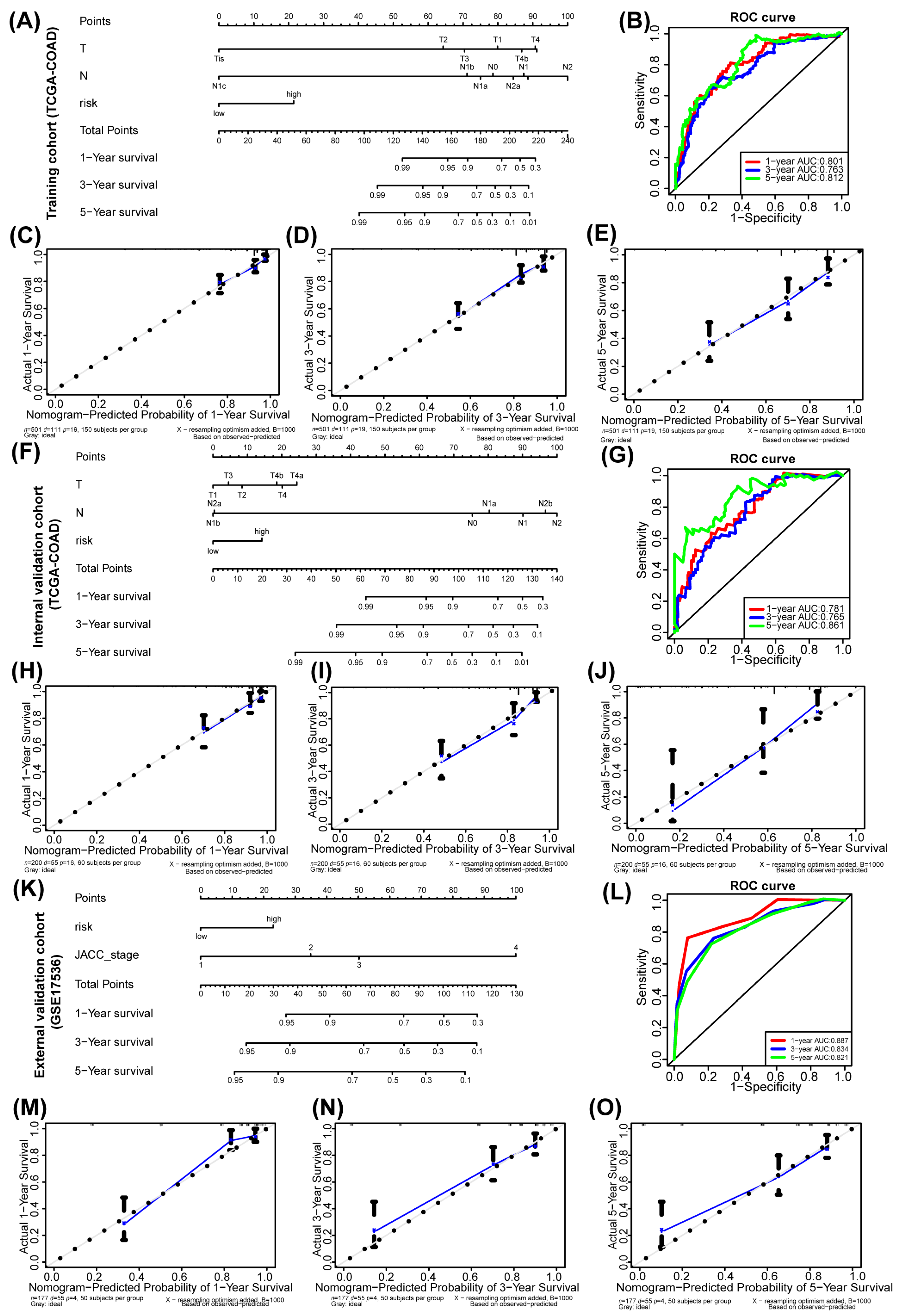
2.5. Nomogram Construction and Conformation
2.6. ScRNA-Seq Data Analysis
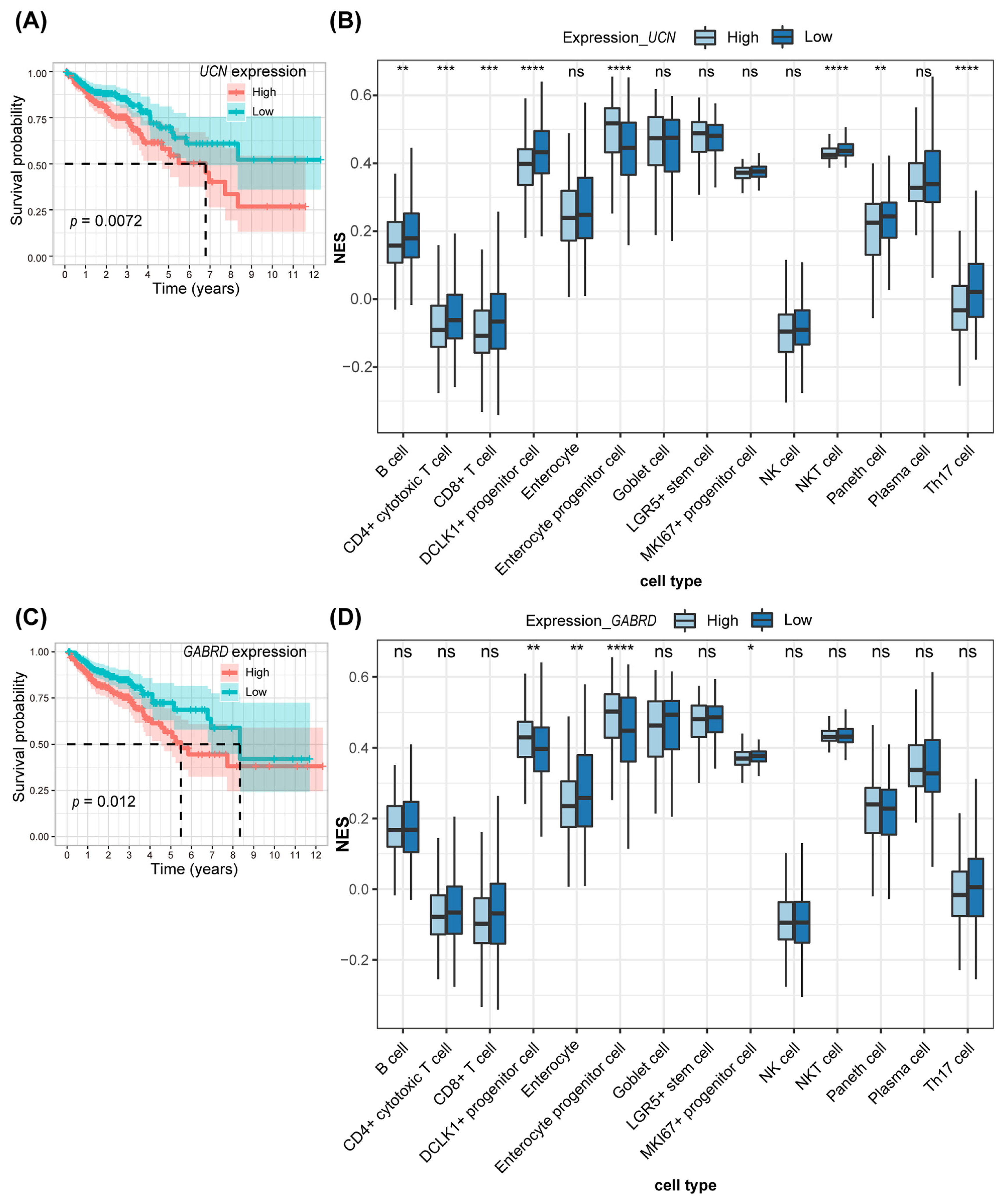
2.7. Immune Cell Infiltration Analysis
2.8. Western Blotting
2.9. RNA Isolation and Quantitative Real-Time PCR
2.10. Cell Viability Assay
2.11. Statistical Analysis
3. Results
3.1. Identification of Survive-Related Gene Modules in CRC Using WGCNA
3.2. Construction of the Prognostic Model Based on the Hub Genes Using LASSO and COX Regression
3.3. Nomogram Prognostic Model Based on Hub Genes
3.4. Immune Infiltration Analysis Combining scRNA-Seq for the Two Key Genes in Prognostic Models
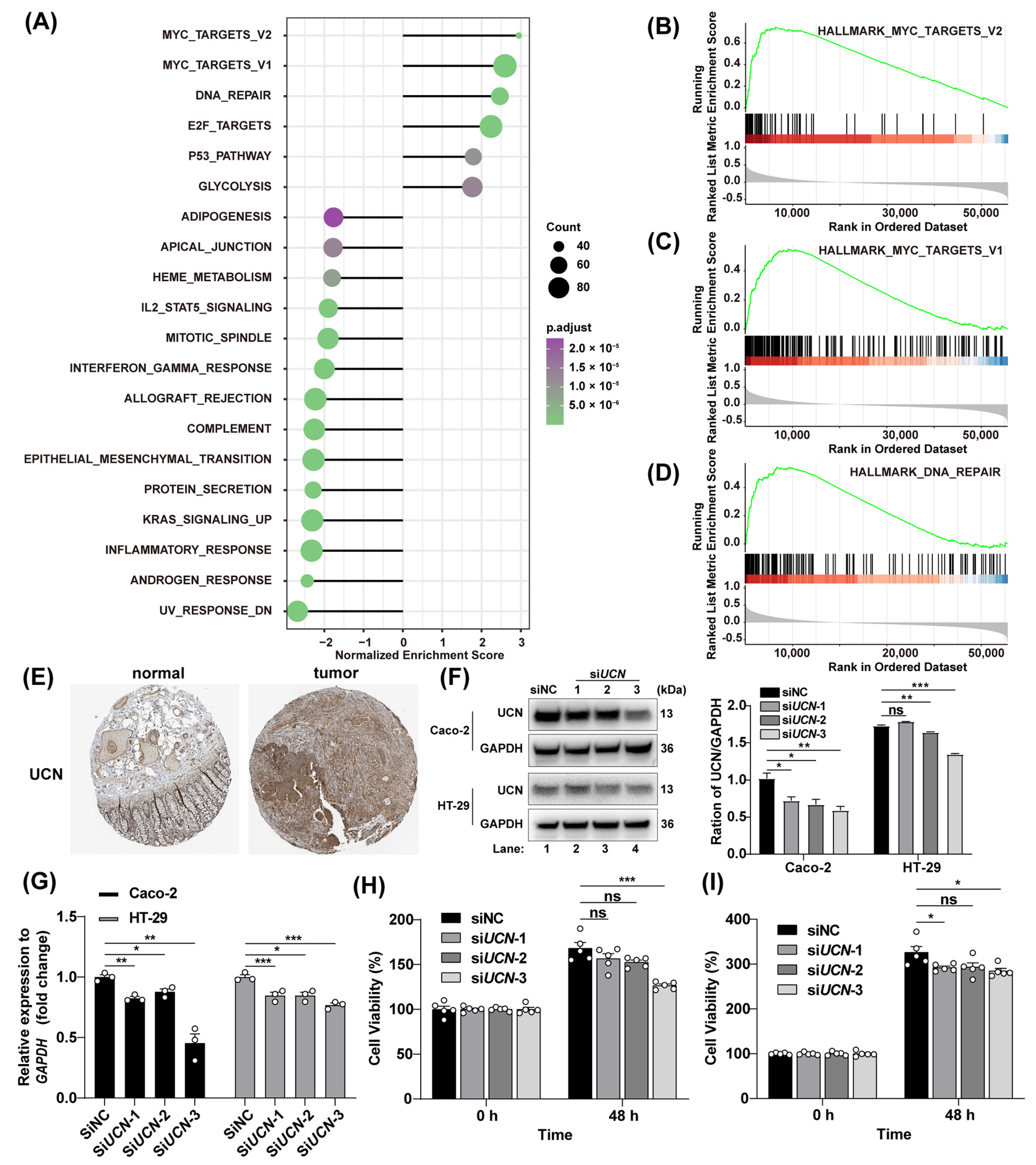
3.5. Tumor-Promoting Action of UCN
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chan, S.C.H.; Liang, J.Q. Advances in tests for colorectal cancer screening and diagnosis. Expert Rev. Mol. Diagn. 2022, 22, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef] [PubMed]
- Simon, K. Colorectal cancer development and advances in screening. Clin. Interv. Aging 2016, 11, 967–976. [Google Scholar] [CrossRef]
- Gupta, S.; Sussman, D.A.; Doubeni, C.A.; Anderson, D.S.; Day, L.; Deshpande, A.R.; Elmunzer, B.J.; Laiyemo, A.O.; Mendez, J.; Somsouk, M.; et al. Challenges and possible solutions to colorectal cancer screening for the underserved. J. Natl. Cancer Inst. 2014, 106, dju032. [Google Scholar] [CrossRef]
- Huang, D.D.; Sun, W.J.; Zhou, Y.W.; Li, P.W.; Chen, F.; Chen, H.W.; Xia, D.J.; Xu, E.P.; Lai, M.D.; Wu, Y.H.; et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018, 37, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, P.; Kolhe, R.; Gahlay, G.K. The clinical relevance of gene expression based prognostic signatures in colorectal cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188513. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Bao, T.Y.; Huang, X.Q.; Lan, Q.W.; Huang, Z.M.; Chen, Y.H.; Hu, Z.D.; Guo, X.G. Machine learning algorithm to construct cuproptosis- and immune-related prognosis prediction model for colon cancer. World J. Gastrointest. Oncol. 2023, 15, 372–388. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, G.; Wang, L.; Liang, M. Exploration and validation of a combined immune and metabolism gene signature for prognosis prediction of colorectal cancer. Front. Endocrinol. 2022, 13, 1069528. [Google Scholar] [CrossRef]
- Huang, X.; Sun, Y.W.; Song, J.; Huang, Y.S.; Shi, H.Z.; Qian, A.H.; Cao, Y.C.; Zhou, Y.C.; Wang, Q.J. Prognostic value of fatty acid metabolism-related genes in colorectal cancer and their potential implications for immunotherapy. Front. Immunol. 2023, 14, 1301452. [Google Scholar] [CrossRef]
- Weng, W.; Zhang, Z.; Huang, W.; Xu, X.; Wu, B.; Ye, T.; Shan, Y.; Shi, K.; Lin, Z. Identification of a competing endogenous RNA network associated with prognosis of pancreatic adenocarcinoma. Cancer Cell Int. 2020, 20, 231. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e3529. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, L.; Qu, J.Z.; Chen, T.T.; Su, Z.J.; Hu, Y.L.; Wang, Y.; Yao, M.D.; Xiao, W.H.; Li, C.; et al. NVD-BM-mediated genetic biosensor triggers accumulation of 7-dehydrocholesterol and inhibits melanoma via Akt1/NF-kB signaling. Aging 2020, 12, 15021–15036. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Mohamed, E.; Sierra, R.A.; Trillo-Tinoco, J.; Cao, Y.; Innamarato, P.; Payne, K.K.; de Mingo Pulido, A.; Mandula, J.; Zhang, S.; Thevenot, P.; et al. The unfolded protein response mediator PERK governs myeloid cell-driven immunosuppression in tumors through inhibition of STING signaling. Immunity 2020, 52, 668–682 e667. [Google Scholar] [CrossRef]
- Kakati, T.; Bhattacharyya, D.K.; Barah, P.; Kalita, J.K. Comparison of methods for differential co-expression analysis for disease biomarker prediction. Comput. Biol. Med. 2019, 113, 103380. [Google Scholar] [CrossRef]
- Kunowska, N.; Rotival, M.; Yu, L.; Choudhary, J.; Dillon, N. Identification of protein complexes that bind to histone H3 combinatorial modifications using super-SILAC and weighted correlation network analysis. Nucleic Acids Res. 2015, 43, 1418–1432. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, J.; Liu, Z.; Liu, X.; Ma, Y.; Zhang, B.; Chen, Y.; Li, X.; Feng, Z.; Yang, N.; et al. Identification of hub genes in colorectal cancer based on weighted gene co-expression network analysis and clinical data from the Cancer Genome Atlas. Biosci. Rep. 2021, 41, BSR20211280. [Google Scholar] [CrossRef]
- Chen, H.; Luo, J.; Guo, J. Development and validation of a five-immune gene prognostic risk model in colon cancer. BMC Cancer 2020, 20, 395. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Fang, Y. Research for expression and prognostic value of GABRD in colon cancer and coexpressed gene network construction based on data mining. Comput. Math. Methods Med. 2021, 2021, 5544182. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Kim, K.Y.; Park, W.C.; Ryu, H.S.; Choi, S.C.; Kim, M.S.; Myung, J.Y.; Choi, H.S.; Kim, E.J.; Lee, M.Y. Enhanced expression of GABRD predicts poor prognosis in patients with colon adenocarcinoma. Transl. Oncol. 2020, 13, 100861. [Google Scholar] [CrossRef]
- Li, X.; Wang, C.Y. From bulk, single-cell to spatial RNA sequencing. Int. J. Oral Sci. 2021, 13, 36. [Google Scholar] [CrossRef]
- Potter, S.S. Single-cell RNA sequencing for the study of development, physiology and disease. Nat. Rev. Nephrol. 2018, 14, 479–492. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Zhong, F.; Chen, Y. UCN-Centric Prognostic Model for Predicting Overall Survival and Immune Response in Colorectal Cancer. Genes 2024, 15, 1139. https://doi.org/10.3390/genes15091139
Liu J, Zhong F, Chen Y. UCN-Centric Prognostic Model for Predicting Overall Survival and Immune Response in Colorectal Cancer. Genes. 2024; 15(9):1139. https://doi.org/10.3390/genes15091139
Chicago/Turabian StyleLiu, Jia, Feiliang Zhong, and Yue Chen. 2024. "UCN-Centric Prognostic Model for Predicting Overall Survival and Immune Response in Colorectal Cancer" Genes 15, no. 9: 1139. https://doi.org/10.3390/genes15091139
APA StyleLiu, J., Zhong, F., & Chen, Y. (2024). UCN-Centric Prognostic Model for Predicting Overall Survival and Immune Response in Colorectal Cancer. Genes, 15(9), 1139. https://doi.org/10.3390/genes15091139